PCA quantification
2016-04-23
Last updated: 2016-04-24
Code version: 74f30e4e4b6c6fea3667336da91b01fb5603564f
library("HDMD")
library("ggplot2")
theme_set(theme_bw(base_size = 12))
library("tidyr")Needs to be run after the [data transformation analysis][data-transformation.html] because it uses the PCA results. We want to demonstrate the effect of our normalization steps on the technical variance as observed in the PCA plots.
Input
Input filtered annotation.
anno_filter <- read.table("../data/annotation-filter.txt", header = TRUE,
stringsAsFactors = FALSE)
head(anno_filter) individual replicate well batch sample_id
1 NA19098 r1 A01 NA19098.r1 NA19098.r1.A01
2 NA19098 r1 A02 NA19098.r1 NA19098.r1.A02
3 NA19098 r1 A04 NA19098.r1 NA19098.r1.A04
4 NA19098 r1 A05 NA19098.r1 NA19098.r1.A05
5 NA19098 r1 A06 NA19098.r1 NA19098.r1.A06
6 NA19098 r1 A07 NA19098.r1 NA19098.r1.A07Input PCA results from [data transformation analysis][data-transformation.html].
pca_raw <- read.table("../data/pca-molecules-filter.txt",
header = TRUE, sep = "\t", stringsAsFactors = FALSE)
pca_cpm <- read.table("../data/pca-molecules-cpm.txt",
header = TRUE, sep = "\t", stringsAsFactors = FALSE)
pca_pois <- read.table("../data/pca-molecules-cpm-trans.txt",
header = TRUE, sep = "\t", stringsAsFactors = FALSE)
pca_final <- read.table("../data/pca-final.txt",
header = TRUE, sep = "\t", stringsAsFactors = FALSE)
stopifnot(pca_raw$Obs.batch == anno_filter$batch,
pca_cpm$Obs.batch == anno_filter$batch,
pca_pois$Obs.batch == anno_filter$batch,
pca_final$Obs.batch == anno_filter$batch)Quantifying PCA results
First visualizing how the pairwise differences between individuals and between replicates of the same individual change after each normalization step.
pca_results <- list(raw = pca_raw[, 3:4],
cpm = pca_cpm[, 3:4],
poisson = pca_pois[, 3:4],
final = pca_final[, 3:4])
distance_results <- NULL
for (analysis in names(pca_results)) {
d <- pairwise.mahalanobis(x = pca_results[[analysis]],
grouping = 1:nrow(pca_results[[analysis]]))$distance
rownames(d) <- anno_filter$sample_id
colnames(d) <- anno_filter$sample_id
d_long <- gather(as.data.frame(d), key = "sample1", value = "distance")
d_long$sample2 <- colnames(d)
stopifnot(d_long$d[d_long$sample1 == d_long$sample2] == 0)
d_long$analysis <- analysis
distance_results <- rbind(distance_results, d_long)
}
distance_results <- separate(distance_results, sample1, into = c("individual1", "replicate1", "well1"), sep = "\\.", remove = FALSE)
distance_results <- separate(distance_results, sample2, into = c("individual2", "replicate2", "well2"), sep = "\\.", remove = FALSE)
# Remove self-comparisons
distance_results <- distance_results[distance_results$sample1 !=
distance_results$sample2, ]
# Classify by comparison
distance_results$same_ind <- distance_results$individual1 == distance_results$individual2
distance_results$same_rep <- distance_results$replicate1 == distance_results$replicate2
distance_results$comparison <- NA
distance_results$comparison[distance_results$same_ind &
distance_results$same_rep] <- "Same individual,\nsame replicate"
distance_results$comparison[distance_results$same_ind &
!distance_results$same_rep] <- "Same individual,\ndifferent replicate"
distance_results$comparison[!distance_results$same_ind] <- "different individual"
stopifnot(!is.na(distance_results$comparison))
table(distance_results$comparison, useNA = "ifany")
different individual
834760
Same individual,\ndifferent replicate
274760
Same individual,\nsame replicate
160608 Order the factors.
distance_results$comparison <- factor(distance_results$comparison,
levels = c("different individual",
"Same individual,\ndifferent replicate",
"Same individual,\nsame replicate"))
distance_results$analysis <- factor(distance_results$analysis,
levels = c("raw", "cpm", "poisson", "final"))Plot.
ggplot(distance_results, aes(x = comparison, y = distance, fill = analysis)) +
geom_boxplot()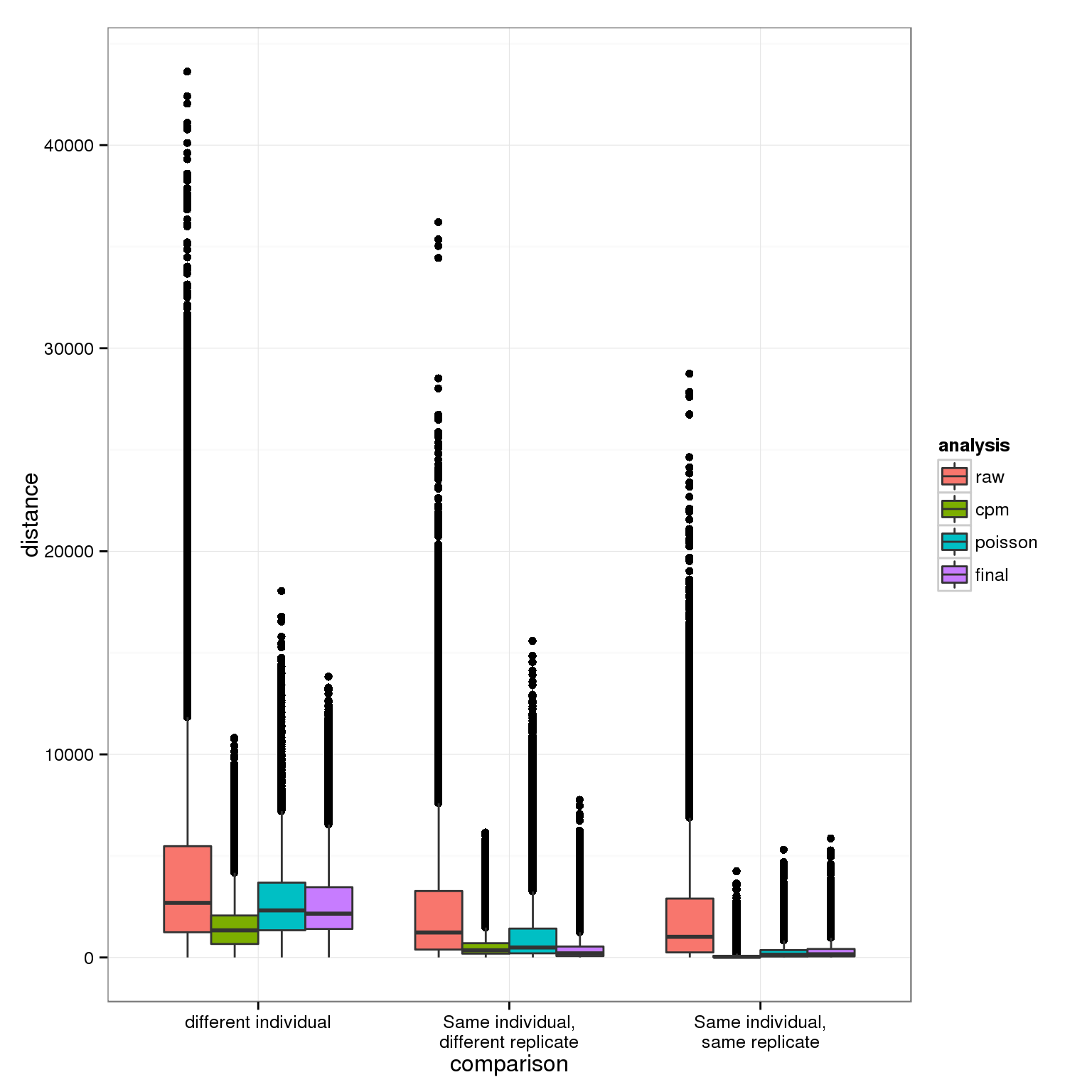
Session information
sessionInfo()R version 3.2.0 (2015-04-16)
Platform: x86_64-unknown-linux-gnu (64-bit)
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] tidyr_0.2.0 ggplot2_1.0.1 HDMD_1.2 MASS_7.3-40 psych_1.5.4
[6] knitr_1.10.5
loaded via a namespace (and not attached):
[1] Rcpp_0.12.0 magrittr_1.5 mnormt_1.5-3 munsell_0.4.2
[5] colorspace_1.2-6 stringr_1.0.0 httr_0.6.1 plyr_1.8.3
[9] tools_3.2.0 parallel_3.2.0 grid_3.2.0 gtable_0.1.2
[13] htmltools_0.2.6 yaml_2.1.13 digest_0.6.8 reshape2_1.4.1
[17] formatR_1.2 bitops_1.0-6 RCurl_1.95-4.6 evaluate_0.7
[21] rmarkdown_0.6.1 labeling_0.3 stringi_0.4-1 scales_0.2.4
[25] proto_0.3-10