Create gene-x-sample count matrices and annotation file
2015-09-30
Last updated: 2016-02-27
Code version: e913fc0a3f954acf436955c670b4cdf1d052c075
library("data.table")
library("dplyr")
library("VennDiagram")This file converts the gene counts results from the analysis pipeline from a sample-by-gene matrix into a standard gene-x-sample matrix. It also creates some annotation files and removes all genes with no observed sequencing data in our experiment.
Specifically, it creates the following files:
- annotation.txt - Annotation file for single cell samples
- annotation-bulk.txt - Annotation file for bulk samples
- reads.txt - Read counts in single cells for all genes with at least one observed read
- reads-bulk.txt - Read counts in bulk samples for all genes with at least one observed read
- molecules.txt - Molecule counts in single cells for all genes with at least one observed read
Read counts for single cell samples
Import raw read counts for single cell samples.
reads_raw <- fread("../data/reads-raw-single-per-sample.txt")
setDF(reads_raw)Create annotation file for single cell samples.
anno <- reads_raw %>%
select(individual:well) %>%
mutate(batch = paste(individual, replicate, sep = "."),
sample_id = paste(batch, well, sep = "."))
head(anno) individual replicate well batch sample_id
1 NA19098 r1 A01 NA19098.r1 NA19098.r1.A01
2 NA19098 r1 A02 NA19098.r1 NA19098.r1.A02
3 NA19098 r1 A03 NA19098.r1 NA19098.r1.A03
4 NA19098 r1 A04 NA19098.r1 NA19098.r1.A04
5 NA19098 r1 A05 NA19098.r1 NA19098.r1.A05
6 NA19098 r1 A06 NA19098.r1 NA19098.r1.A06Transpose the matrix so that it is gene-x-sample.
reads <- reads_raw %>%
select(starts_with("ENSG"), starts_with("ERCC")) %>%
t
colnames(reads) <- anno$sample_id
reads[1:5, 1:5] NA19098.r1.A01 NA19098.r1.A02 NA19098.r1.A03
ENSG00000186092 0 0 0
ENSG00000237683 0 0 0
ENSG00000235249 0 0 0
ENSG00000185097 0 0 0
ENSG00000269831 0 0 0
NA19098.r1.A04 NA19098.r1.A05
ENSG00000186092 0 0
ENSG00000237683 42 0
ENSG00000235249 0 0
ENSG00000185097 0 0
ENSG00000269831 0 0Molecule counts for single cell samples
Import raw molecule counts for single cell samples.
molecules_raw <- fread("../data/molecules-raw-single-per-sample.txt")
setDF(molecules_raw)Confirm single cell samples in reads and molecules files are in the same order.
stopifnot(anno$individual == molecules_raw$individual,
anno$replicate == molecules_raw$replicate,
anno$well == molecules_raw$well)Transpose the matrix so that it is gene-x-sample.
molecules <- molecules_raw %>%
select(starts_with("ENSG"), starts_with("ERCC")) %>%
t
colnames(molecules) <- anno$sample_id
molecules[1:5, 1:5] NA19098.r1.A01 NA19098.r1.A02 NA19098.r1.A03
ENSG00000186092 0 0 0
ENSG00000237683 0 0 0
ENSG00000235249 0 0 0
ENSG00000185097 0 0 0
ENSG00000269831 0 0 0
NA19098.r1.A04 NA19098.r1.A05
ENSG00000186092 0 0
ENSG00000237683 1 0
ENSG00000235249 0 0
ENSG00000185097 0 0
ENSG00000269831 0 0Read counts for bulk samples
Import raw read counts for bulk samples.
reads_bulk_raw <- fread("../data/reads-raw-bulk-per-sample.txt")
setDF(reads_bulk_raw)Create annotation file for bulk samples.
anno_bulk <- reads_bulk_raw %>%
select(individual:well) %>%
mutate(batch = paste(individual, replicate, sep = "."),
sample_id = paste(batch, well, sep = "."))
head(anno_bulk) individual replicate well batch sample_id
1 NA19098 r1 bulk NA19098.r1 NA19098.r1.bulk
2 NA19098 r2 bulk NA19098.r2 NA19098.r2.bulk
3 NA19098 r3 bulk NA19098.r3 NA19098.r3.bulk
4 NA19101 r1 bulk NA19101.r1 NA19101.r1.bulk
5 NA19101 r2 bulk NA19101.r2 NA19101.r2.bulk
6 NA19101 r3 bulk NA19101.r3 NA19101.r3.bulkTranspose the matrix so that it is gene-x-sample.
reads_bulk <- reads_bulk_raw %>%
select(starts_with("ENSG"), starts_with("ERCC")) %>%
t
colnames(reads_bulk) <- anno_bulk$sample_id
reads_bulk[1:5, 1:5] NA19098.r1.bulk NA19098.r2.bulk NA19098.r3.bulk
ENSG00000186092 0 0 0
ENSG00000237683 50 61 39
ENSG00000235249 0 0 0
ENSG00000185097 0 0 0
ENSG00000269831 0 0 0
NA19101.r1.bulk NA19101.r2.bulk
ENSG00000186092 0 0
ENSG00000237683 56 41
ENSG00000235249 0 0
ENSG00000185097 0 0
ENSG00000269831 0 0Observed genes
Not all of the 20419 genes were observed in the experiment.
stopifnot(rownames(reads_bulk) == rownames(reads),
rownames(reads) == rownames(molecules))
genes_observed_reads_bulk <- rownames(reads_bulk)[rowSums(reads_bulk) > 0]
genes_observed_reads <- rownames(reads)[rowSums(reads) > 0]
genes_observed_molecules <- rownames(molecules)[rowSums(molecules) > 0]
stopifnot(genes_observed_molecules %in% genes_observed_reads)18726 genes had at least one observation in the single cell read data, 18726 genes had at least one observation in the single cell molecule data, and 18056 genes had at least one observation in the bulk read data. As expected, all genes with at least one observed molecule in at least one single cell also had at least one observed read in at least one single cell.
genes_venn <- venn.diagram(x = list("reads" = genes_observed_reads,
"molecules" = genes_observed_molecules,
"reads bulk" = genes_observed_reads_bulk),
filename = NULL, euler.d = FALSE, scaled = FALSE)
grid.newpage()
grid.draw(genes_venn)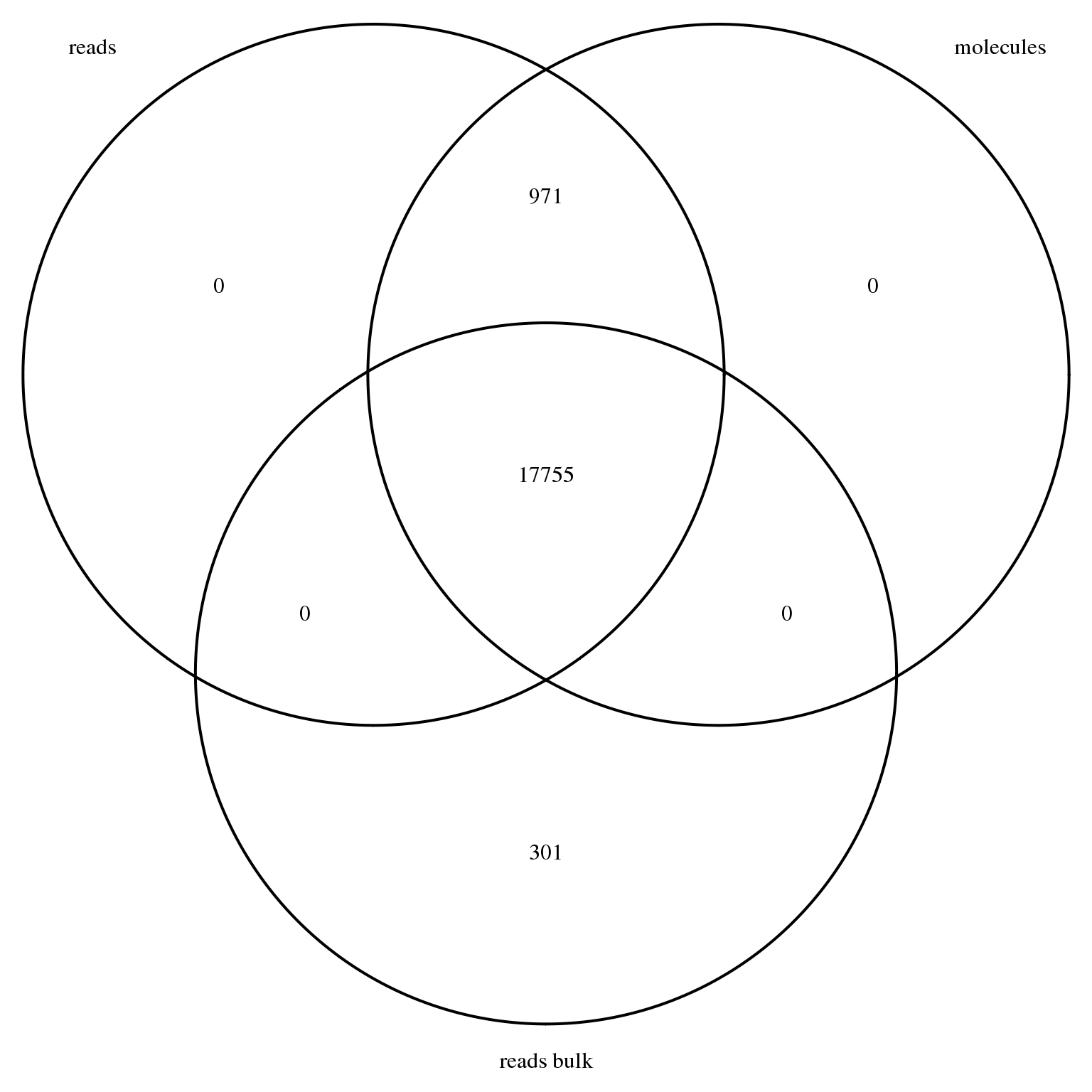
We remove all genes with no observed read in either the bulk or single cell samples.
genes_observed <- union(genes_observed_reads, genes_observed_reads_bulk)
reads <- reads[rownames(reads) %in% genes_observed, ]
molecules <- molecules[rownames(molecules) %in% genes_observed, ]
reads_bulk <- reads_bulk[rownames(reads_bulk) %in% genes_observed, ]This leaves a total of 19027 genes.
Write files
Output annotation files.
write.table(anno, "../data/annotation.txt", quote = FALSE, sep = "\t",
row.names = FALSE)
write.table(anno_bulk, "../data/annotation-bulk.txt", quote = FALSE, sep = "\t",
row.names = FALSE)Output read counts.
write.table(reads, "../data/reads.txt", quote = FALSE, sep = "\t",
col.names = NA)
write.table(reads_bulk, "../data/reads-bulk.txt", quote = FALSE, sep = "\t",
col.names = NA)Output molecule counts.
write.table(molecules, "../data/molecules.txt", quote = FALSE, sep = "\t",
col.names = NA)Session information
sessionInfo()R version 3.2.0 (2015-04-16)
Platform: x86_64-unknown-linux-gnu (64-bit)
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] grid stats graphics grDevices utils datasets methods
[8] base
other attached packages:
[1] VennDiagram_1.6.9 dplyr_0.4.2 data.table_1.9.4 knitr_1.10.5
loaded via a namespace (and not attached):
[1] Rcpp_0.12.0 magrittr_1.5 R6_2.1.1 stringr_1.0.0
[5] httr_0.6.1 plyr_1.8.3 tools_3.2.0 parallel_3.2.0
[9] DBI_0.3.1 htmltools_0.2.6 lazyeval_0.1.10 yaml_2.1.13
[13] digest_0.6.8 assertthat_0.1 reshape2_1.4.1 formatR_1.2
[17] bitops_1.0-6 RCurl_1.95-4.6 evaluate_0.7 rmarkdown_0.6.1
[21] stringi_0.4-1 chron_2.3-45