Coefficient of variation
Joyce Hsiao
2015-09-26
Last updated: 2015-12-11
Code version: 90871a71ff87e914ca4a695037b835130cd57dab
Objectives
We used Kruskal-Wallis test to compare coefficient of variation between individual (3 coefficient of variations from the batches for per individual). The p-value distribution of the per gene Kruskal-Wallis test does not resemeble p-value distribution of the commonly know per gene tests of differential gene expression.
Visulization of the count distributions of the top 9 genes that are different in coefficient of variation between individuals using the Kruskal-Wallis test did not reveal systematic different between the count distributions.
We then applied Kwame Okrah’s HTShape package to compute skewness and kurtoss estimtes of each gene with each unique batches, and also across the three individuals. The SO-plot shows that there are a good proportion of the genes that are skewed in their count distribution.
Set up
library(ggplot2)
library(edgeR)
library(limma)Prepare data
Input annotation
anno_single <- read.table("../data/annotation-filter.txt", header = TRUE,
stringsAsFactors = FALSE)
head(anno_single) individual replicate well batch sample_id
1 NA19098 r1 A01 NA19098.r1 NA19098.r1.A01
2 NA19098 r1 A02 NA19098.r1 NA19098.r1.A02
3 NA19098 r1 A04 NA19098.r1 NA19098.r1.A04
4 NA19098 r1 A05 NA19098.r1 NA19098.r1.A05
5 NA19098 r1 A06 NA19098.r1 NA19098.r1.A06
6 NA19098 r1 A07 NA19098.r1 NA19098.r1.A07Input molecule counts
molecules <- read.table("../data/molecules-final.txt", header = TRUE, stringsAsFactors = FALSE)Compute coefficient of variation
Per individual coefficient of variation.
molecules_cv <-
lapply(1:length(unique(anno_single$individual)), function(per_person) {
molecules_per_person <-
molecules[ , anno_single$individual == unique(anno_single$individual)[per_person]]
cv_foo <- data.frame(mean = apply(molecules_per_person, 1, mean, na.rm = TRUE),
cv = apply(molecules_per_person, 1, sd, na.rm = TRUE) /
apply(molecules_per_person, 1, mean, na.rm = TRUE) )
rownames(cv_foo) <- rownames(molecules)
cv_foo <- cv_foo[rowSums(is.na(cv_foo)) == 0, ]
cv_foo$individual <- unique(anno_single$individual)[per_person]
return(cv_foo)
})
names(molecules_cv) <- unique(anno_single$individual)Per batch coefficient of variation.
batch <- anno_single$batch
molecules_cv_batch <-
lapply(1:length(unique(batch)), function(per_batch) {
molecules_per_batch <- molecules[ , batch == unique(batch)[per_batch]]
cv_foo <- data.frame(mean = apply(molecules_per_batch, 1, mean, na.rm = TRUE),
cv = apply(molecules_per_batch, 1, sd, na.rm = TRUE) /
apply(molecules_per_batch, 1, mean, na.rm = TRUE) )
rownames(cv_foo) <- rownames(molecules)
cv_foo <- cv_foo[rowSums(is.na(cv_foo)) == 0, ]
cv_foo$batch <- unique(batch)[per_batch]
return(cv_foo)
})
names(molecules_cv_batch) <- unique(batch)Subset CV data to include only genes that have data across all batches.
all_genes <- lapply(1:3, function(ii) {
if (ii == 1) {
return(intersect(intersect(rownames(molecules_cv_batch[[ 1+ (ii-1)*3]]),
rownames(molecules_cv_batch[[ 2+ (ii-1)*3]]) ),
rownames(molecules_cv_batch[[3 + (ii-1)*3]]) ) )
}
if (ii == 2) {
return(
intersect(rownames(molecules_cv_batch[[ 1+ (ii-1)*3]]),
rownames(molecules_cv_batch[[ 2+ (ii-1)*3]]) ) )
}
if (ii == 3) {
return(
intersect(intersect(rownames(molecules_cv_batch[[ 1+ (ii-1)*3 - 1]]),
rownames(molecules_cv_batch[[ 2+ (ii-1)*3 - 1]]) ),
rownames(molecules_cv_batch[[3 + (ii-1)*3 - 1]]) ) )
}
})
all_genes <- intersect(intersect(all_genes[[1]], all_genes[[2]]), all_genes[[3]])
str(all_genes) chr [1:10564] "ENSG00000237683" "ENSG00000188976" ...molecules_cv_batch <-
lapply(1: length(molecules_cv_batch), function(per_batch) {
ii_include <- which(rownames(molecules_cv_batch[[per_batch]]) %in% all_genes)
molecules_cv_batch[[per_batch]][ ii_include, ]
})
nrow(molecules_cv_batch[[1]])[1] 10564Kruskal-Wallis test
A nonparametric test commonly used for comparison of multiple groups that have a very small sample size.
df_cv <- lapply(1:nrow(molecules_cv_batch[[1]]), function(per_gene) {
do.call(rbind, lapply(1: length(molecules_cv_batch), function(per_batch) {
molecules_cv_batch[[per_batch]][per_gene, ] })
)
})
# Make individual factor variable
individual <-
do.call( c,
lapply( strsplit(df_cv[[1]]$batch, fixed = TRUE, split = "."),
"[[", 1) )
# Kruskal walls
kw_pval <- lapply(1:nrow(molecules_cv_batch[[1]]), function(per_gene) {
result <- kruskal.test(cv ~ factor(individual),
data = df_cv[[per_gene]])
data.frame(pval = result$p.value,
gene = rownames(molecules_cv_batch[[1]])[per_gene])
})
kw_pval <- do.call(rbind, kw_pval)
summary(kw_pval$pval) Min. 1st Qu. Median Mean 3rd Qu. Max.
0.04394 0.11940 0.24940 0.34890 0.56580 0.98620 ggplot(data = kw_pval, aes(x = pval)) + geom_histogram() +
ggtitle("Kruskal-Wallis p-value")stat_bin: binwidth defaulted to range/30. Use 'binwidth = x' to adjust this.
Top 9 in Kruskal-Wallis
ii_diff <- order(kw_pval$pval)[1:9]
gene_subset <- df_cv[ii_diff]
df_cv_merge <- lapply(1:9, function(per_gene) {
data.frame(gene_subset[[per_gene]],
individual = factor(individual),
gene = kw_pval$gene[ii_diff[per_gene]])
})
df_cv_merge <- do.call(rbind, df_cv_merge)
ggplot(df_cv_merge, aes( x = factor(individual), y = cv)) +
geom_point(aes(col = factor(individual))) + facet_grid(. ~ gene)
batch <- anno_single$batch
count_cell <- lapply(1:length(unique(batch)), function(per_batch) {
ii_batch <- which(batch %in% unique(batch)[per_batch])
ii_gene <- which( rownames(molecules) %in% kw_pval$gene[ii_diff] )
df_foo <- molecules[ii_gene, ii_batch]
colnames(df_foo) <- ""
# df_meta <- data.frame(df_foo,
# individual = anno_single$individual[ii_batch][1],
# batch = anno_single$batch[ii_batch][1],
# genes = rownames(df_foo))
# df_meta
as.matrix(df_foo)
})
names(count_cell) <- unique(batch)
for (i in 1:length(count_cell)) {
par(mfrow=c(3,3))
for (j in 1:length(count_cell)) {
plot(density(count_cell[[j]][i, ]),
main = names(count_cell)[[j]],
xlab = "Molecule count")
}
}







Explore quantiles
Use Kwame’s package to compare shapes of distributions.
require(devtools)Loading required package: devtools#install_github("kokrah/HTshape")
require(HTShape)Loading required package: HTShapeSelect genes that have valid CV measures in all batches.
SO-plot for one batch…
ii_genes <- which(rownames(molecules) %in% all_genes )
batch <- anno_single$batch
fit_shape <- lapply(1:length(unique(batch)), function(per_batch) {
ii_batch <- which(batch == unique(batch)[per_batch])
count_foo <- as.matrix(molecules[ii_genes, ii_batch])
shape_results <- fitShape( log(count_foo + 1) )
list(L3 = shape_results$lrats[ "LR3", ],
L4 = shape_results$lrats[ "LR4", ] )
})
plotSO(fit_shape[[1]]$L3, fit_shape[[1]]$L4)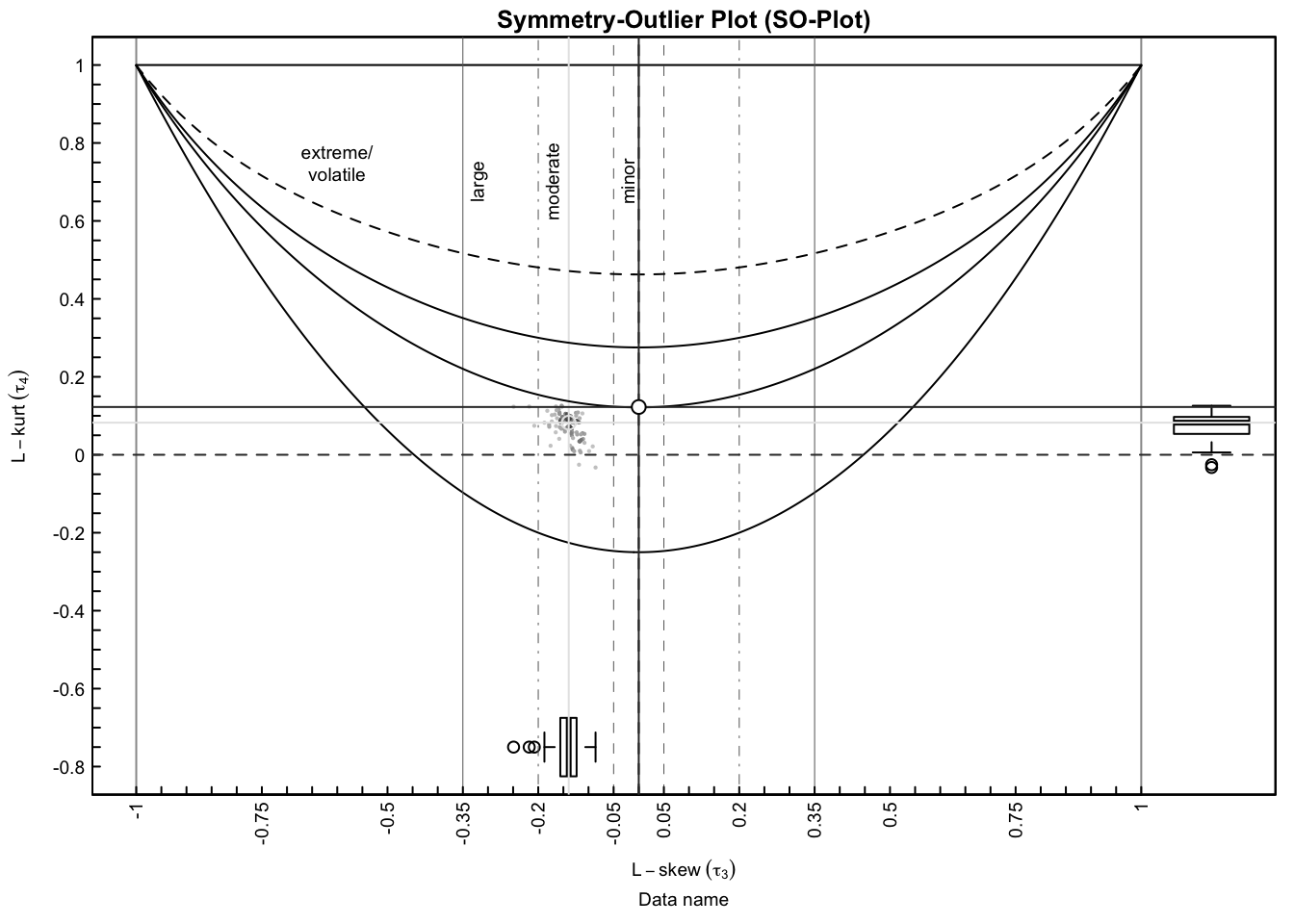
SO-plot for the entire data set.
ii_genes <- which(rownames(molecules) %in% all_genes )
fit_shape <- fitShape( log(molecules[ii_genes, ] + 1) )
plotSO(fit_shape$lrats[1, ], fit_shape$lrats[2, ])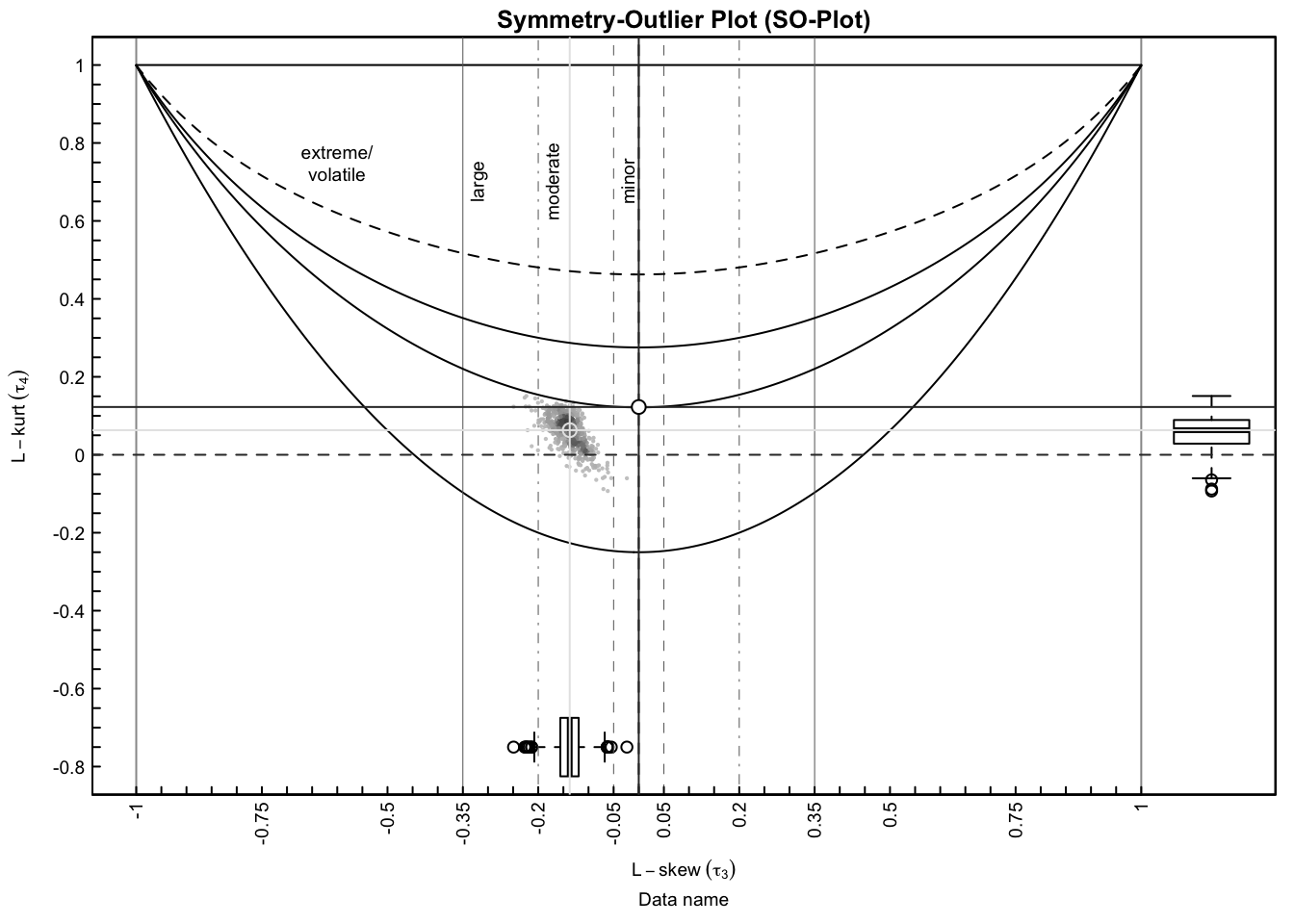
Session information
sessionInfo()R version 3.2.1 (2015-06-18)
Platform: x86_64-apple-darwin13.4.0 (64-bit)
Running under: OS X 10.10.5 (Yosemite)
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] HTShape_1.0.0 devtools_1.9.1 edgeR_3.10.5 limma_3.24.15
[5] ggplot2_1.0.1 knitr_1.11
loaded via a namespace (and not attached):
[1] Rcpp_0.12.2 magrittr_1.5 MASS_7.3-45
[4] munsell_0.4.2 colorspace_1.2-6 stringr_1.0.0
[7] plyr_1.8.3 tools_3.2.1 grid_3.2.1
[10] gtable_0.1.2 KernSmooth_2.23-15 htmltools_0.2.6
[13] yaml_2.1.13 digest_0.6.8 reshape2_1.4.1
[16] formatR_1.2.1 memoise_0.2.1 evaluate_0.8
[19] rmarkdown_0.8.1 labeling_0.3 stringi_1.0-1
[22] scales_0.3.0 proto_0.3-10