PCA
John Blischak
2017-08-14
Last updated: 2018-01-29
Code version: f6b7f76
Batch 1 only
Setup
library("cowplot")
library("dplyr")
library("edgeR")
library("ggplot2")
library("stringr")
library("tidyr")
theme_set(theme_cowplot())
source("../code/functions.R")
library("Biobase") # has to be loaded last to use `combine`Import data.
eset <- readRDS("../data/eset.rds")
esetExpressionSet (storageMode: lockedEnvironment)
assayData: 54792 features, 4992 samples
element names: exprs
protocolData: none
phenoData
sampleNames: 03162017-A01 03162017-A02 ... 12142017-H12 (4992
total)
varLabels: experiment well ... valid_id (40 total)
varMetadata: labelDescription
featureData
featureNames: ENSG00000000003 ENSG00000000005 ... WBGene00235374
(54792 total)
fvarLabels: chr start ... source (6 total)
fvarMetadata: labelDescription
experimentData: use 'experimentData(object)'
Annotation: Limit this analysis to batch 1 since it deals with the spike-ins.
eset <- eset[, eset$batch == "b1"]
dim(eset)Features Samples
54792 960 Remove samples with bad cell number or TRA-1-60.
eset_quality <- eset[, eset$cell_number == 1 & eset$tra1.60]
dim(eset_quality)Features Samples
54792 869 Separate by source.
eset_ce <- eset_quality[fData(eset_quality)$source == "C. elegans", ]
head(featureNames(eset_ce))[1] "WBGene00000001" "WBGene00000002" "WBGene00000003" "WBGene00000004"
[5] "WBGene00000005" "WBGene00000006"eset_dm <- eset_quality[fData(eset_quality)$source == "D. melanogaster", ]
head(featureNames(eset_dm))[1] "FBgn0000008" "FBgn0000014" "FBgn0000015" "FBgn0000017" "FBgn0000018"
[6] "FBgn0000022"eset_ercc <- eset_quality[fData(eset_quality)$source == "ERCC",
eset_quality$ERCC != "Not added"]
head(featureNames(eset_ercc))[1] "ERCC-00002" "ERCC-00003" "ERCC-00004" "ERCC-00009" "ERCC-00012"
[6] "ERCC-00013"eset_hs <- eset_quality[fData(eset_quality)$source == "H. sapiens", ]
head(featureNames(eset_hs))[1] "ENSG00000000003" "ENSG00000000005" "ENSG00000000419" "ENSG00000000457"
[5] "ENSG00000000460" "ENSG00000000938"Define a function for filtering by percentage of cells in which a gene is detected.
present <- function(x, percent = 0.50) mean(x > 0) >= percentERCC
Remove zeros.
eset_ercc_clean <- eset_ercc[rowSums(exprs(eset_ercc)) != 0, ]
dim(eset_ercc_clean)Features Samples
88 347 Only keep genes which are observed in at least 50% of the samples.
eset_ercc_clean <- eset_ercc_clean[apply(exprs(eset_ercc_clean), 1, present), ]
dim(eset_ercc_clean)Features Samples
34 347 mol_ercc_cpm <- cpm(exprs(eset_ercc_clean), log = TRUE)
hist(rowMeans(mol_ercc_cpm))
pca_ercc <- run_pca(mol_ercc_cpm)
plot_pca(pca_ercc$PCs, pcx = 1, pcy = 2, explained = pca_ercc$explained,
metadata = pData(eset_ercc_clean),
color = "experiment")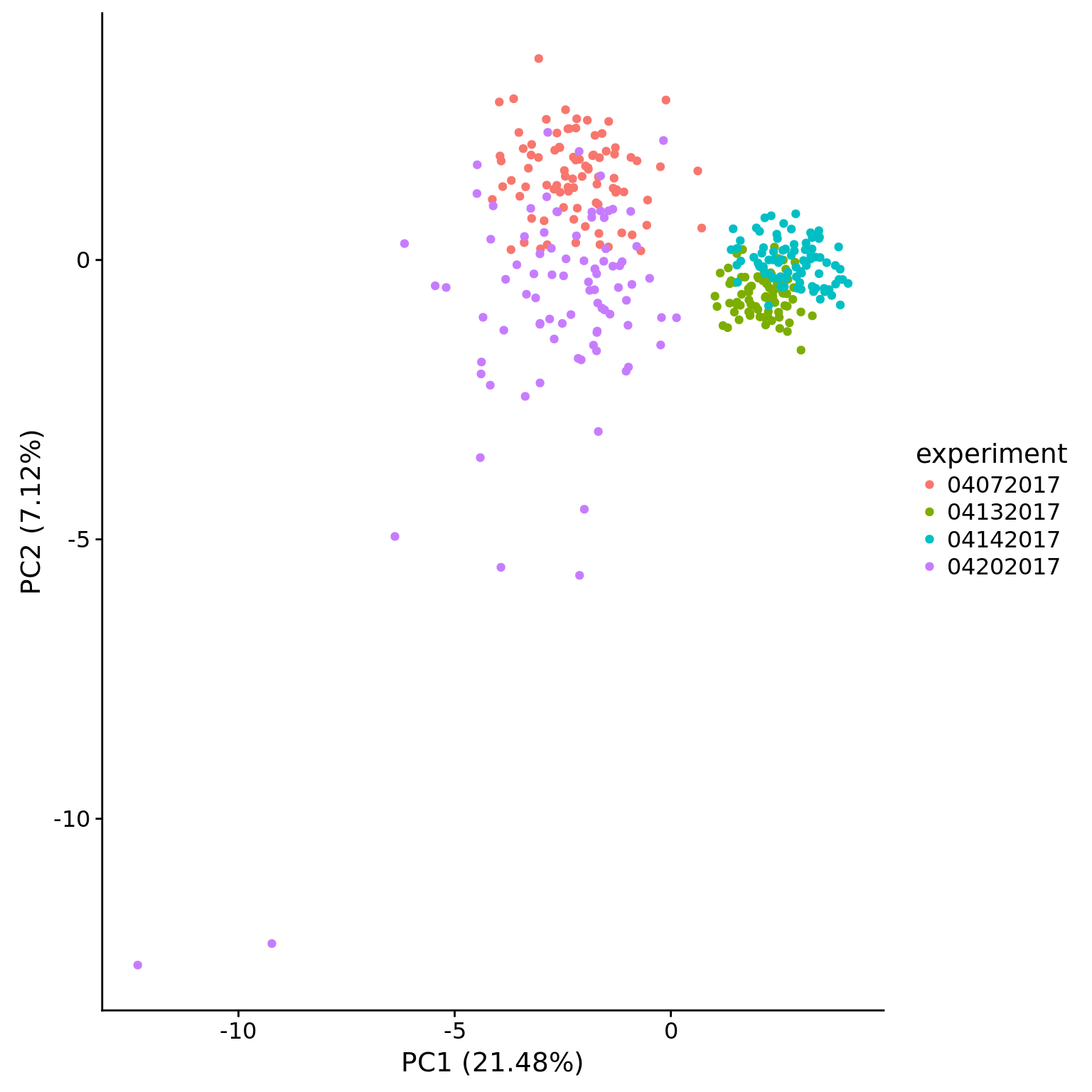
plot_pca(pca_ercc$PCs, pcx = 3, pcy = 4, explained = pca_ercc$explained,
metadata = pData(eset_ercc_clean),
color = "experiment")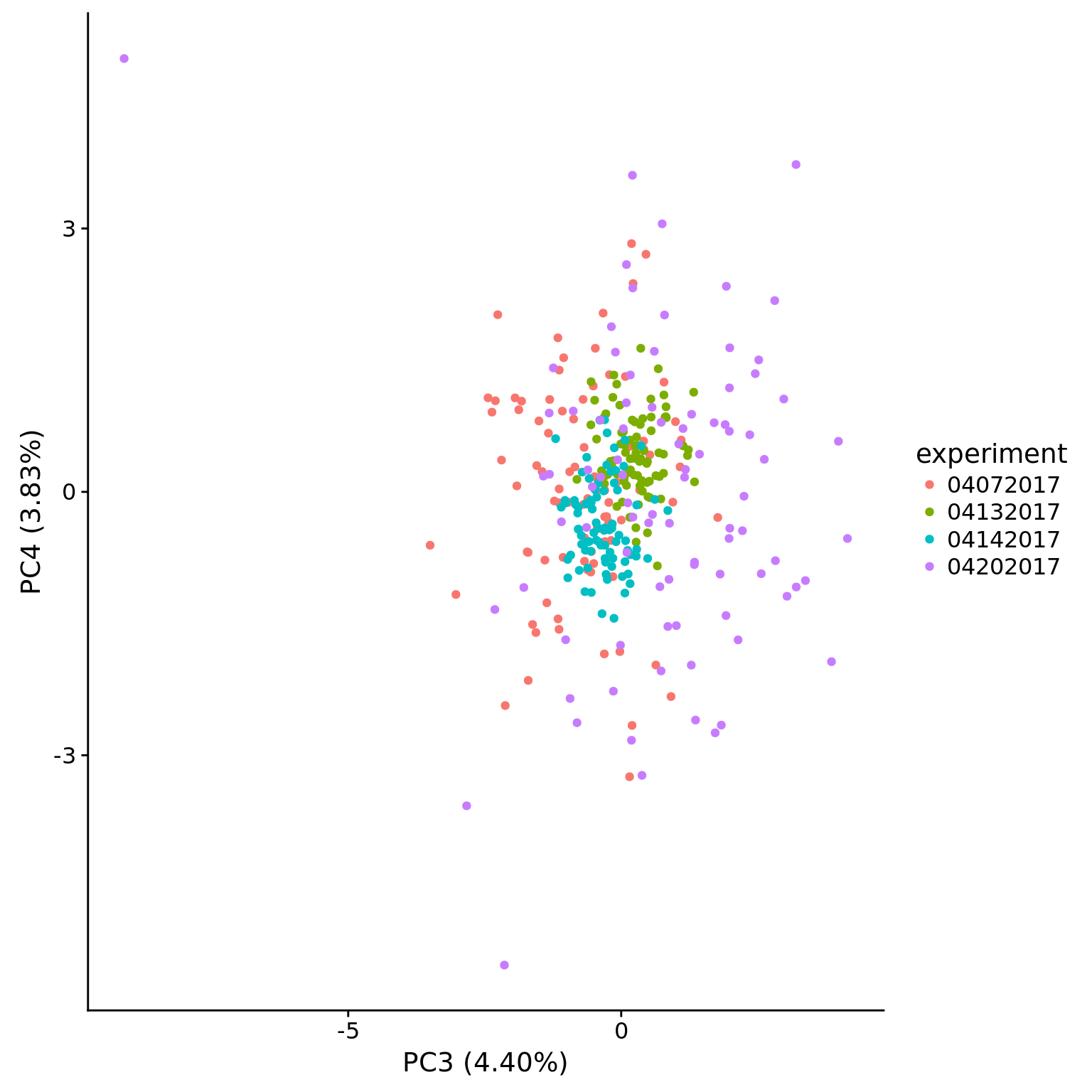
plot_pca(pca_ercc$PCs, pcx = 5, pcy = 6, explained = pca_ercc$explained,
metadata = pData(eset_ercc_clean),
color = "experiment")
plot_pca(pca_ercc$PCs, pcx = 5, pcy = 6, explained = pca_ercc$explained,
metadata = pData(eset_ercc_clean),
color = "ERCC")
Drosophila
Remove zeros.
eset_dm_clean <- eset_dm[rowSums(exprs(eset_dm)) != 0, ]
dim(eset_dm_clean)Features Samples
11561 869 Only keep genes which are observed in at least 50% of the samples.
eset_dm_clean <- eset_dm_clean[apply(exprs(eset_dm_clean), 1, present), ]
dim(eset_dm_clean)Features Samples
327 869 Convert to log2 counts per million.
mol_dm_cpm <- cpm(exprs(eset_dm_clean), log = TRUE)
hist(rowMeans(mol_dm_cpm))
pca_dm <- run_pca(mol_dm_cpm)
plot_pca(pca_dm$PCs, pcx = 1, pcy = 2, explained = pca_dm$explained,
metadata = pData(eset_dm_clean),
color = "experiment")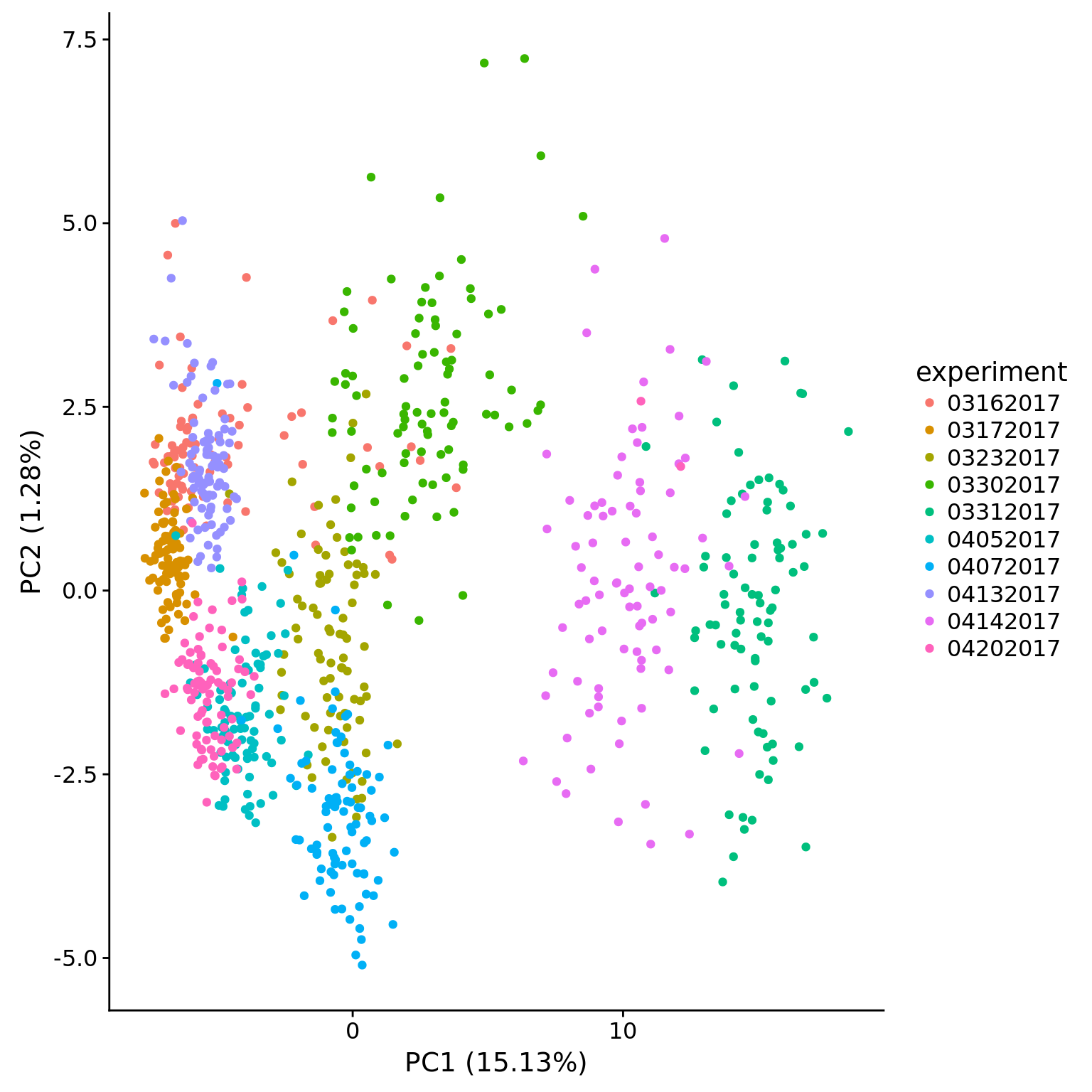
plot_pca(pca_dm$PCs, pcx = 1, pcy = 2, explained = pca_dm$explained,
metadata = pData(eset_dm_clean),
color = "fly", factors = "fly")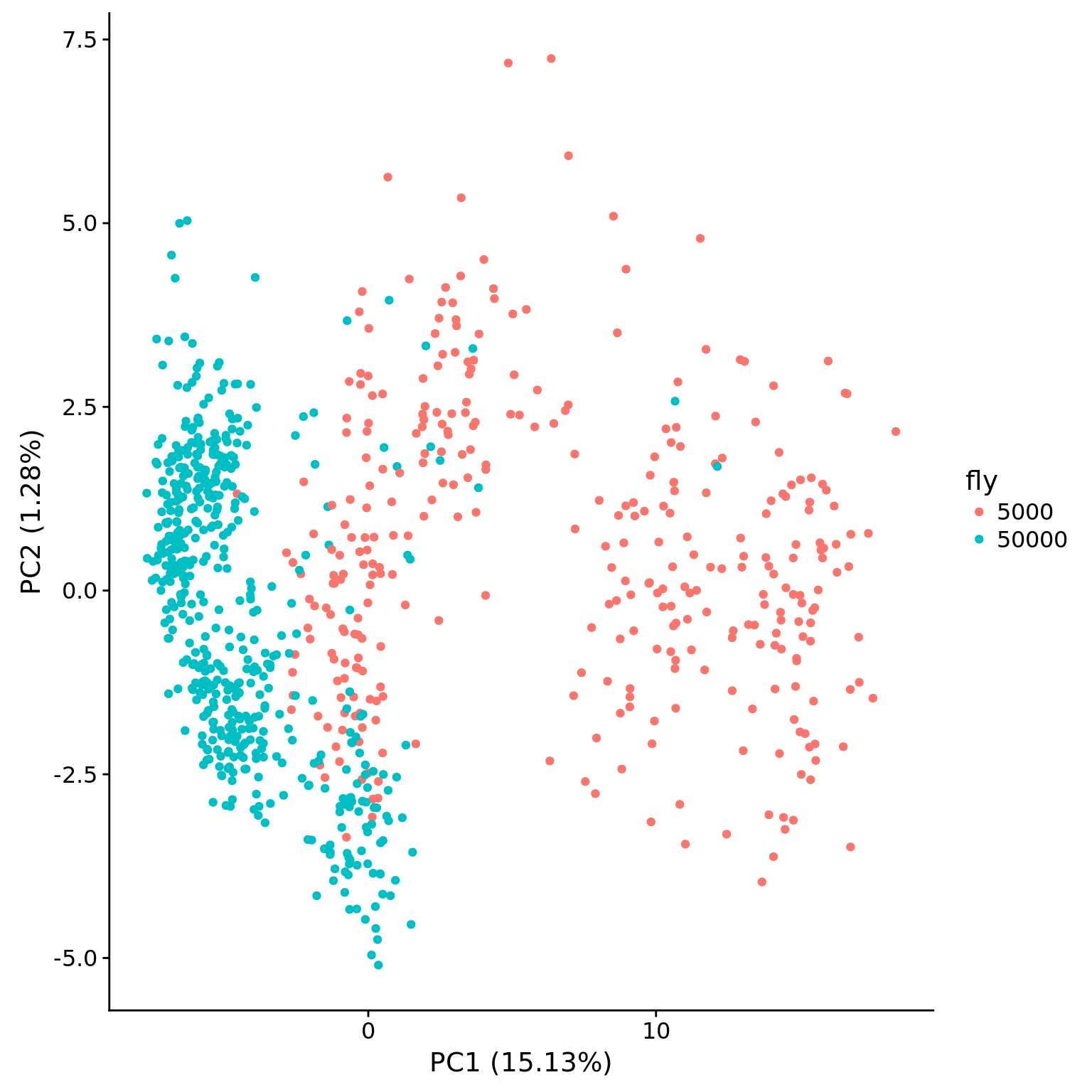
Drosophila - 5 pg
Select only samples that received 5 pg.
eset_dm_5pg <- eset_dm[, eset_dm$fly == 5000]
dim(eset_dm_5pg)Features Samples
13832 343 Remove zeros.
eset_dm_5pg_clean <- eset_dm_5pg[rowSums(exprs(eset_dm_5pg)) != 0, ]
dim(eset_dm_5pg_clean)Features Samples
9833 343 Only keep genes which are observed in at least 50% of the samples.
eset_dm_5pg_clean <- eset_dm_5pg_clean[apply(exprs(eset_dm_5pg_clean), 1, present), ]
dim(eset_dm_5pg_clean)Features Samples
154 343 Convert to log2 counts per million.
mol_dm_cpm_5pg <- cpm(exprs(eset_dm_5pg_clean), log = TRUE)
hist(rowMeans(mol_dm_cpm_5pg))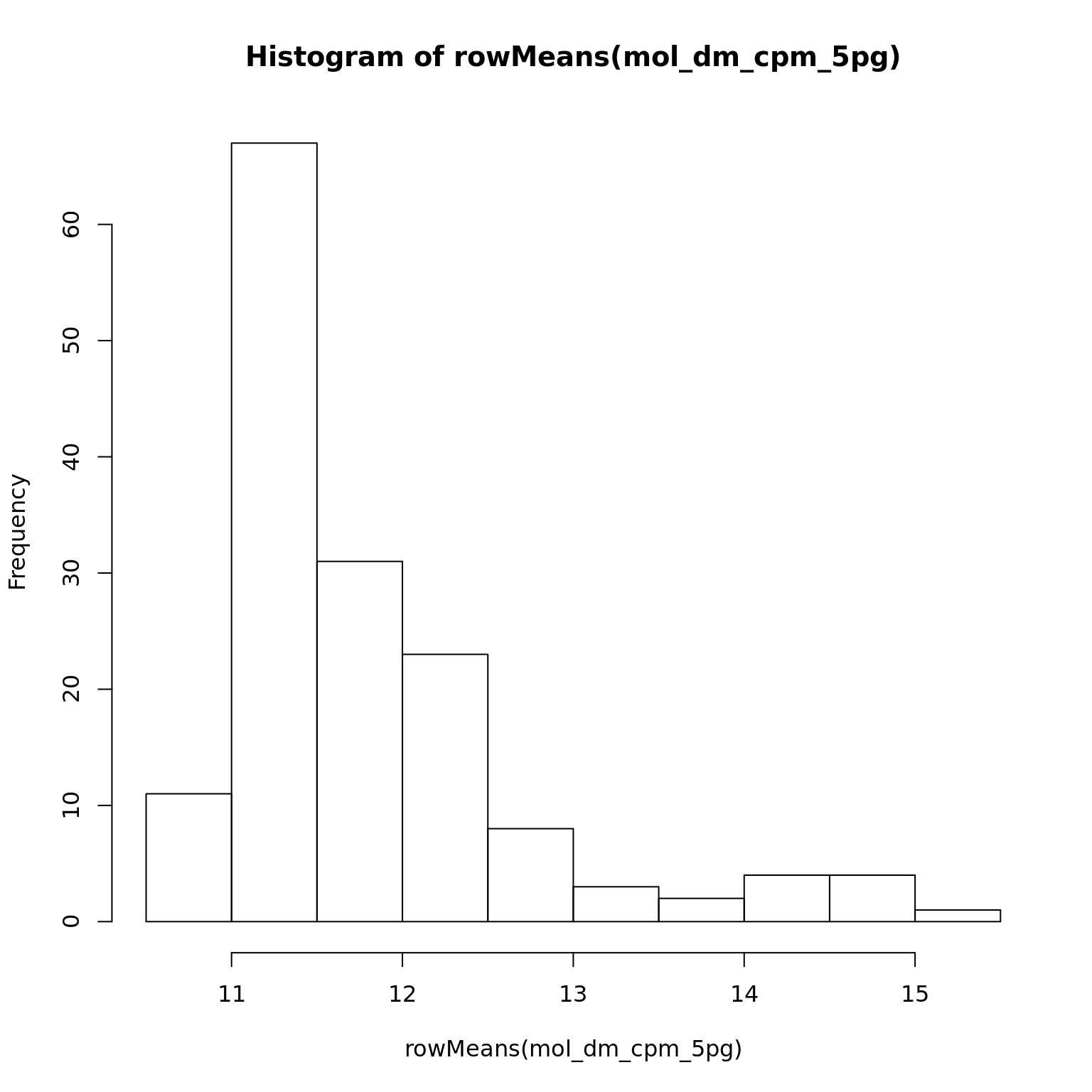
pca_dm_5pg <- run_pca(mol_dm_cpm_5pg)
plot_pca(pca_dm_5pg$PCs, pcx = 1, pcy = 2, explained = pca_dm_5pg$explained,
metadata = pData(eset_dm_5pg_clean),
color = "experiment")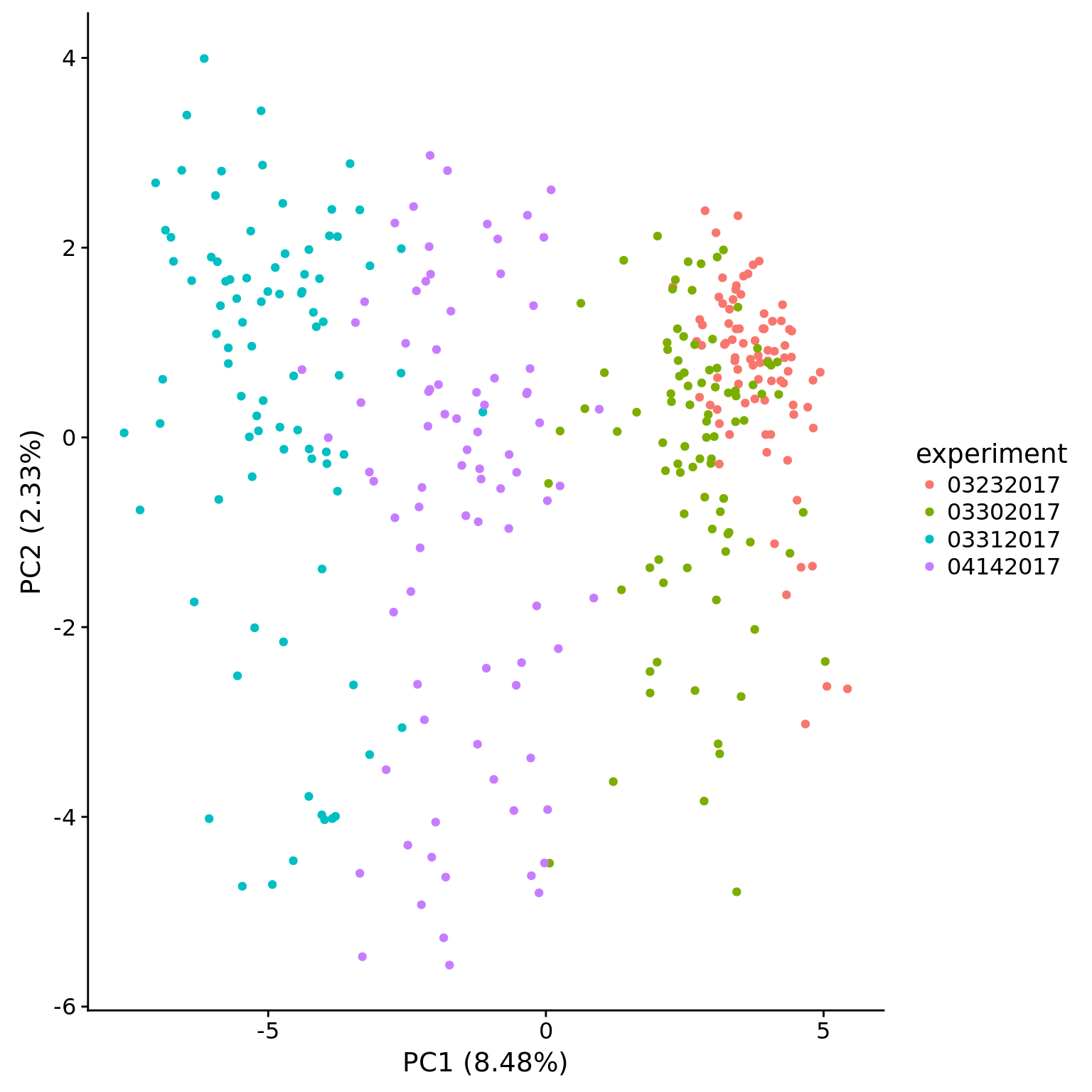
plot_pca(pca_dm_5pg$PCs, pcx = 3, pcy = 4, explained = pca_dm_5pg$explained,
metadata = pData(eset_dm_5pg_clean),
color = "experiment")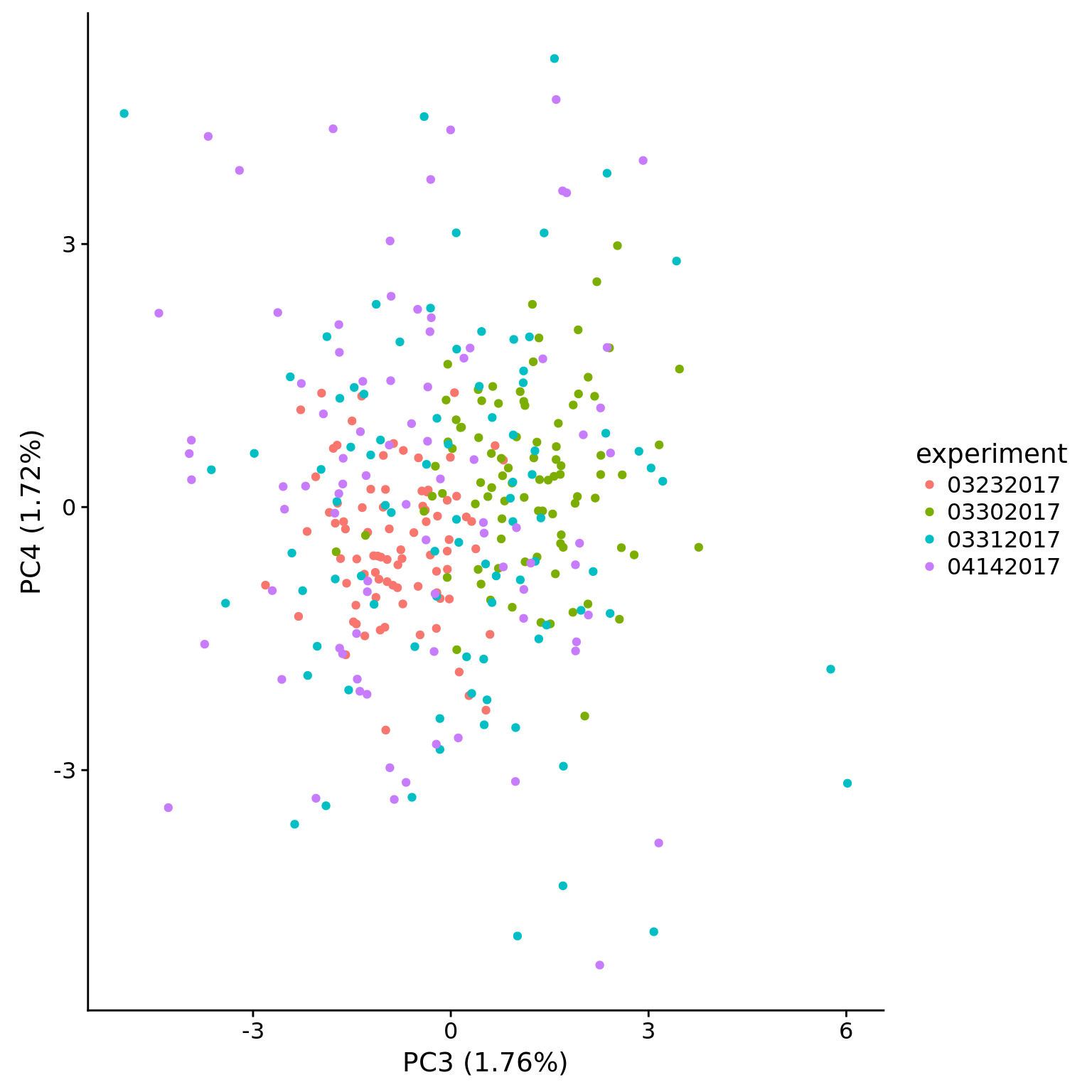
Drosophila - 50 pg
Select only samples that received 50 pg.
eset_dm_50pg <- eset_dm[, eset_dm$fly == 50000]
dim(eset_dm_50pg)Features Samples
13832 526 Remove zeros.
eset_dm_50pg_clean <- eset_dm_50pg[rowSums(exprs(eset_dm_50pg)) != 0, ]
dim(eset_dm_50pg_clean)Features Samples
11120 526 Only keep genes which are observed in at least 50% of the samples.
eset_dm_50pg_clean <- eset_dm_50pg_clean[apply(exprs(eset_dm_50pg_clean), 1, present), ]
dim(eset_dm_50pg_clean)Features Samples
530 526 Convert to log2 counts per million.
mol_dm_cpm_50pg <- cpm(exprs(eset_dm_50pg_clean), log = TRUE)
hist(rowMeans(mol_dm_cpm_50pg))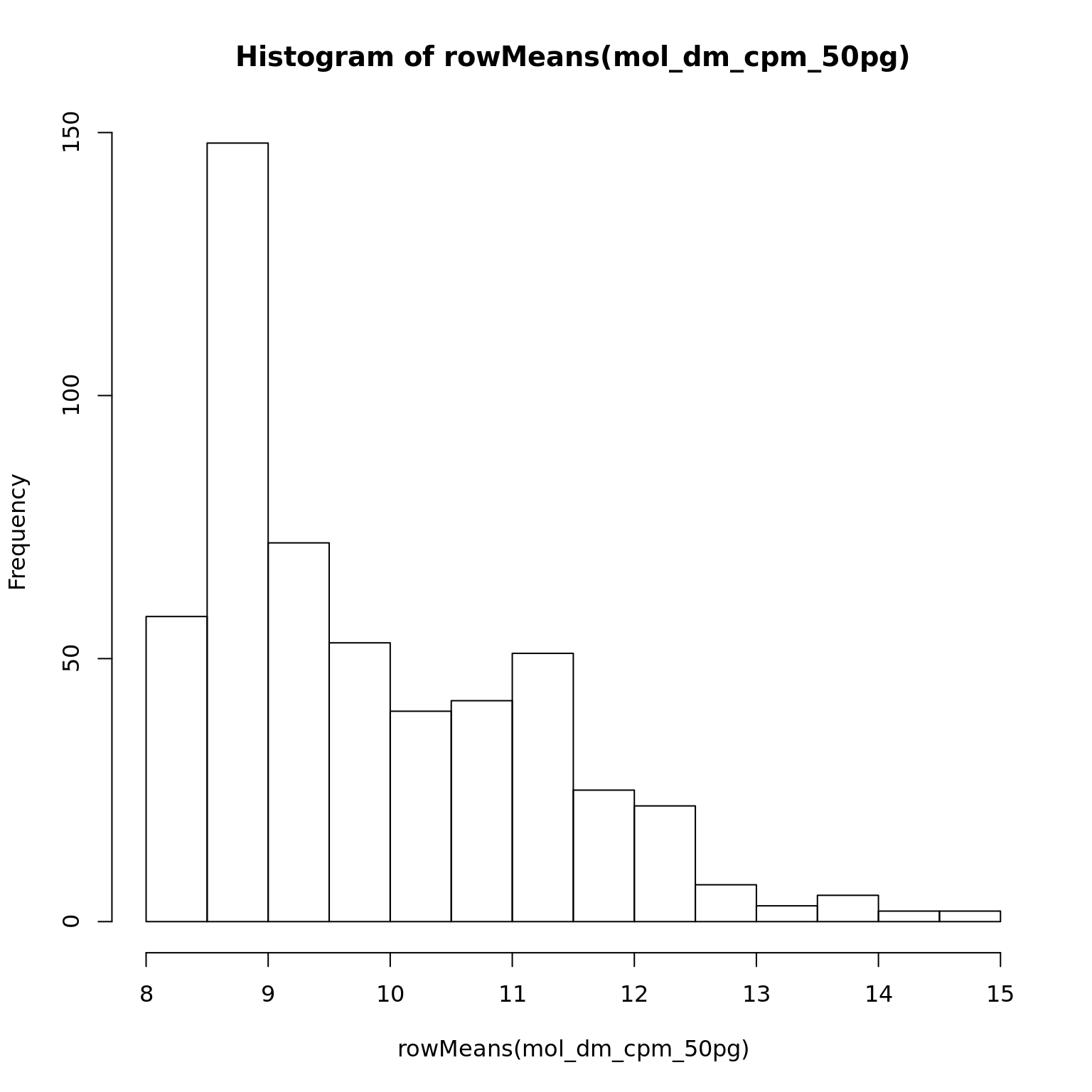
pca_dm_50pg <- run_pca(mol_dm_cpm_50pg)
plot_pca(pca_dm_50pg$PCs, pcx = 1, pcy = 2, explained = pca_dm_50pg$explained,
metadata = pData(eset_dm_50pg_clean),
color = "experiment")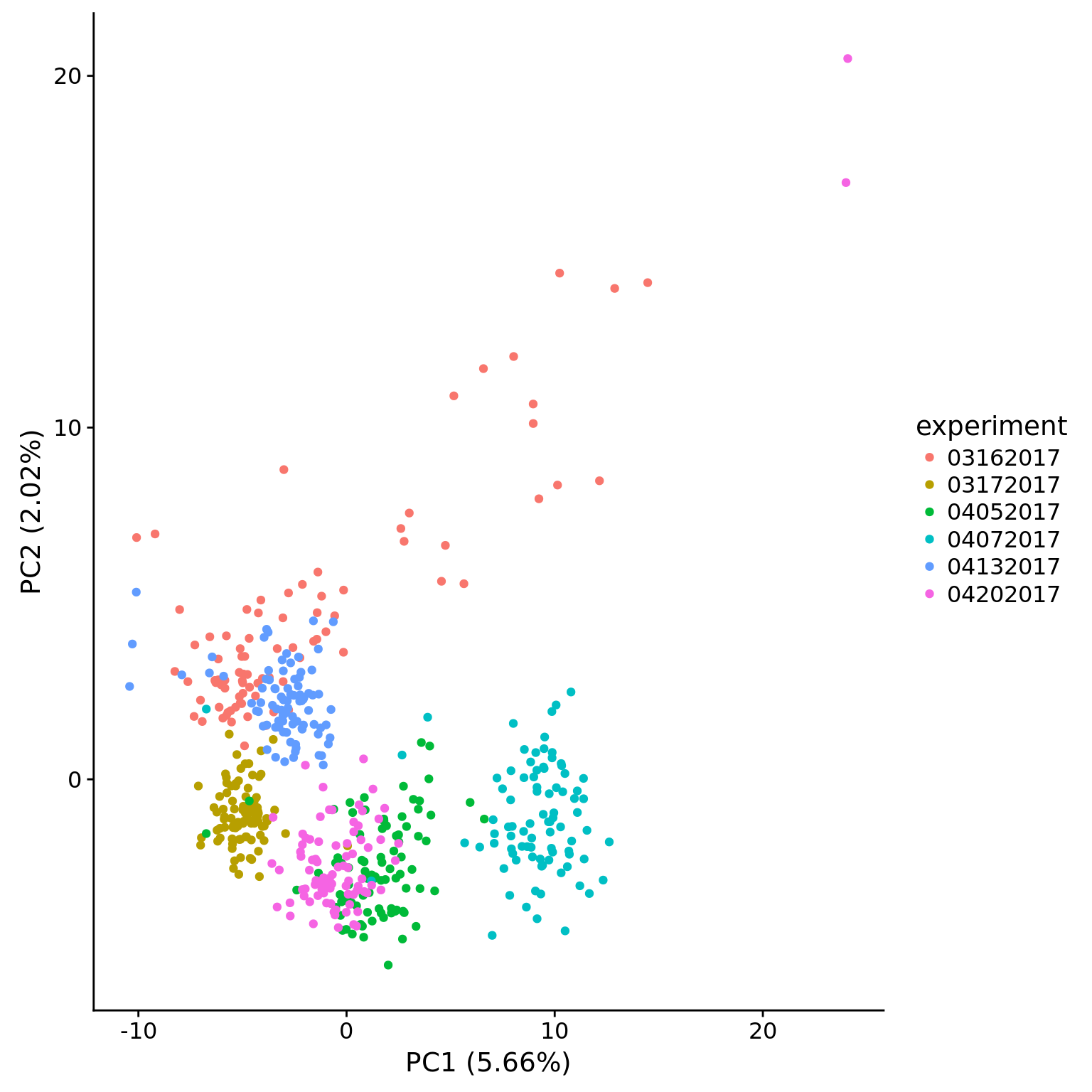
plot_pca(pca_dm_50pg$PCs, pcx = 3, pcy = 4, explained = pca_dm_50pg$explained,
metadata = pData(eset_dm_50pg_clean),
color = "experiment")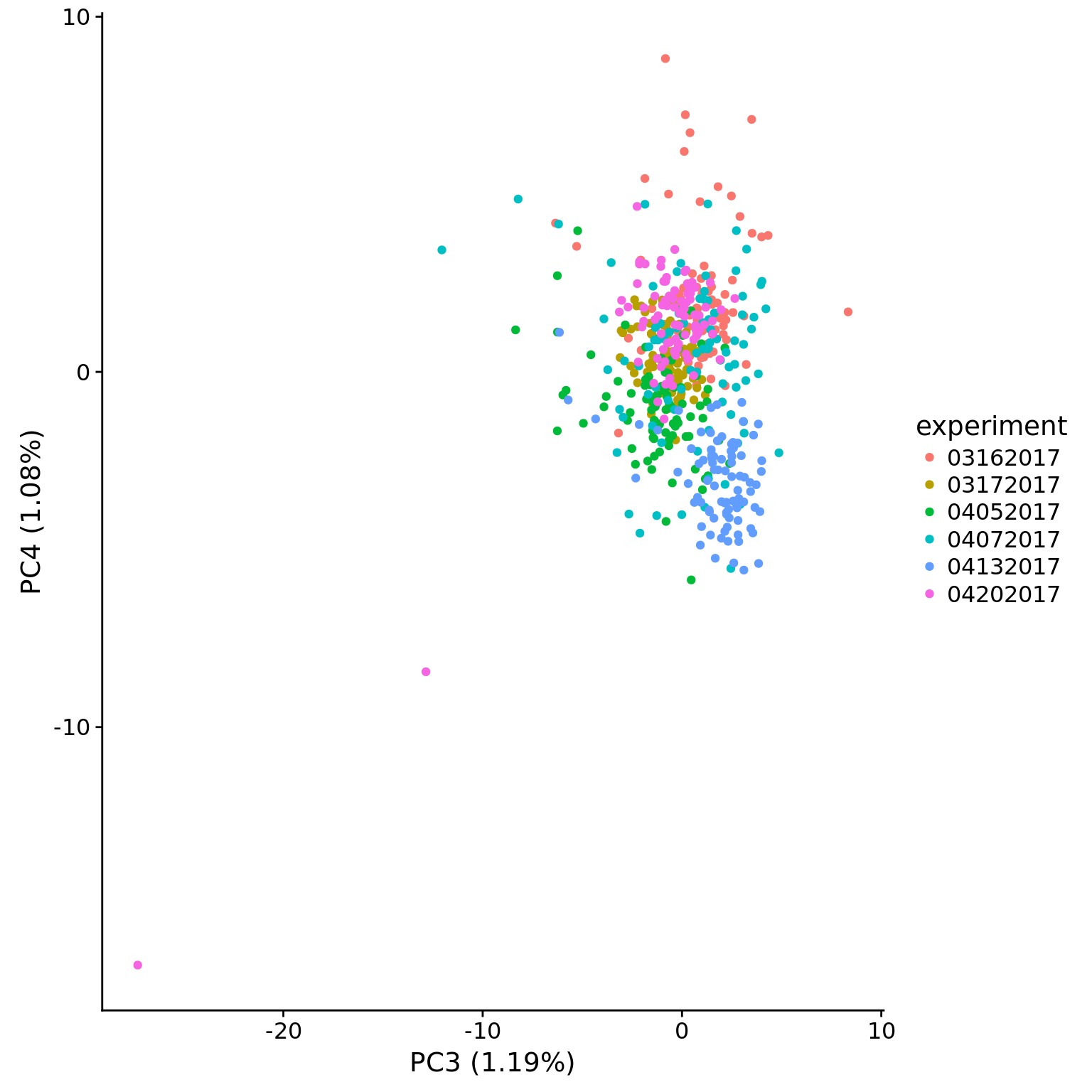
plot_pca(pca_dm_50pg$PCs, pcx = 5, pcy = 6, explained = pca_dm_50pg$explained,
metadata = pData(eset_dm_50pg_clean),
color = "experiment")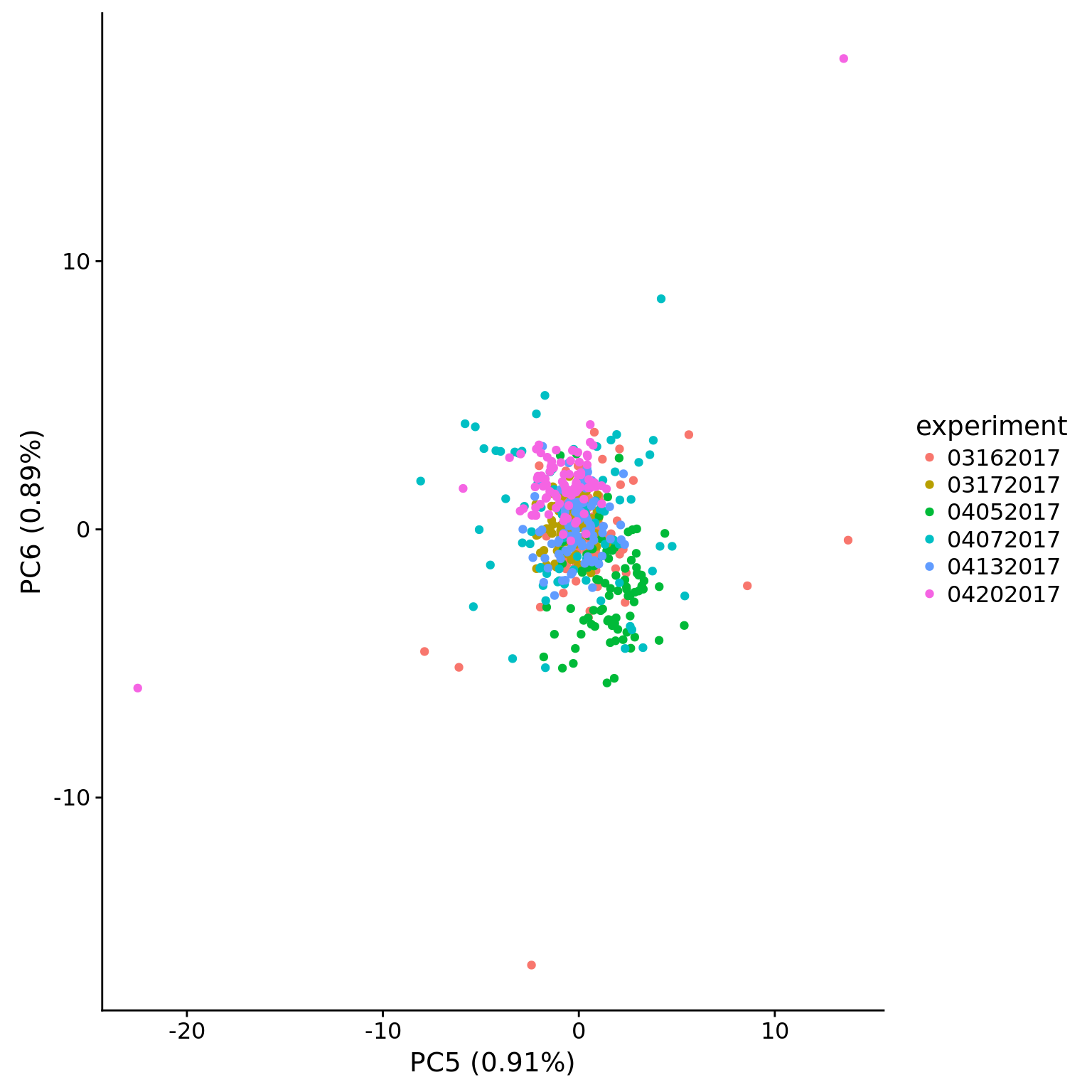
C. elegans
Remove zeros.
eset_ce_clean <- eset_ce[rowSums(exprs(eset_ce)) != 0, ]
dim(eset_ce_clean)Features Samples
13105 869 Only keep genes which are observed in at least 50% of the samples.
eset_ce_clean <- eset_ce_clean[apply(exprs(eset_ce_clean), 1, present), ]
dim(eset_ce_clean)Features Samples
10 869 Convert to log2 counts per million.
mol_ce_cpm <- cpm(exprs(eset_ce_clean), log = TRUE)
# Remove samples with no observations for this subset
zeros_ce <- colSums(exprs(eset_ce_clean)) > 0
eset_ce_clean <- eset_ce_clean[, zeros_ce]
mol_ce_cpm <- mol_ce_cpm[, zeros_ce]
hist(rowMeans(mol_ce_cpm))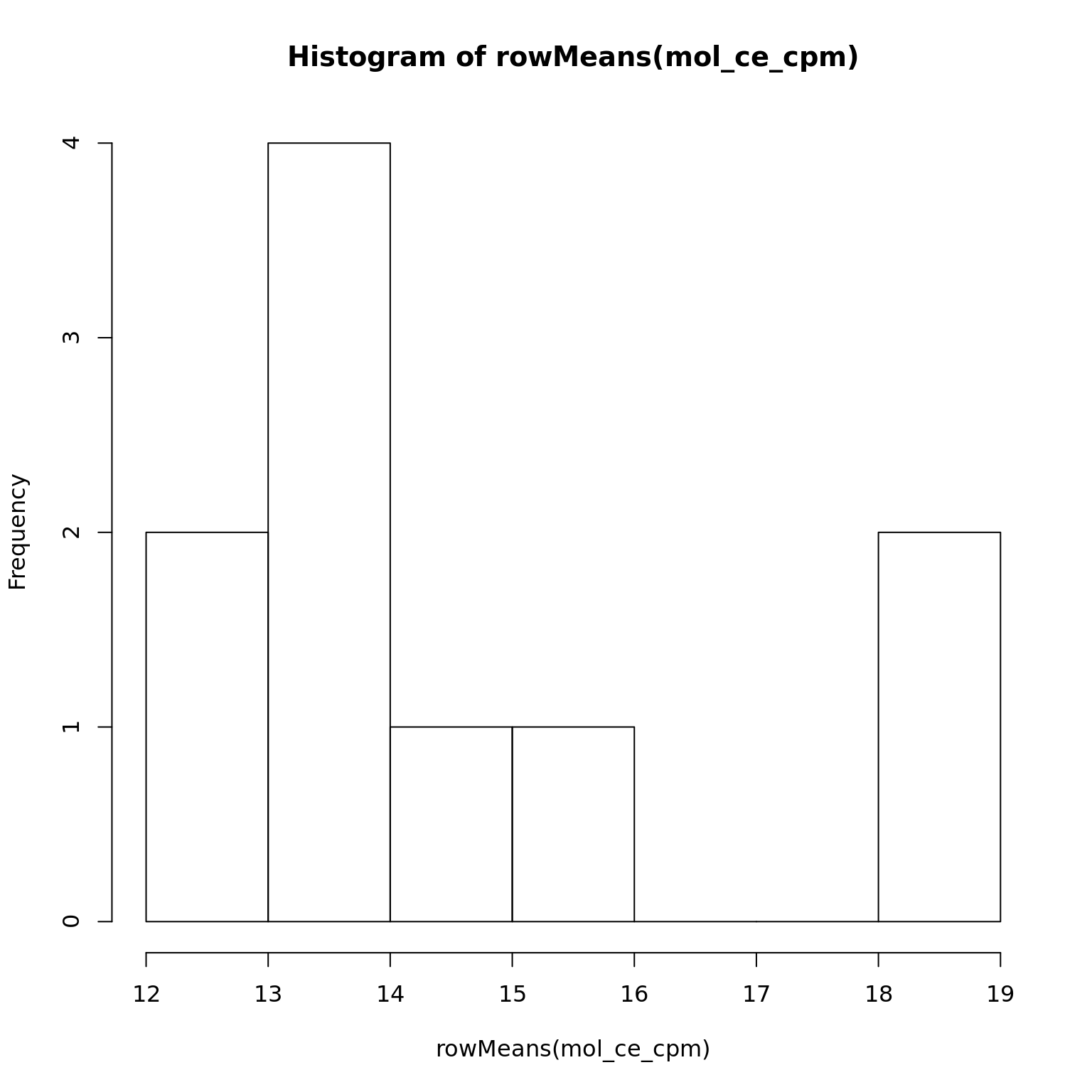
pca_ce <- run_pca(mol_ce_cpm)
plot_pca(pca_ce$PCs, pcx = 1, pcy = 2, explained = pca_ce$explained,
metadata = pData(eset_ce_clean),
color = "experiment")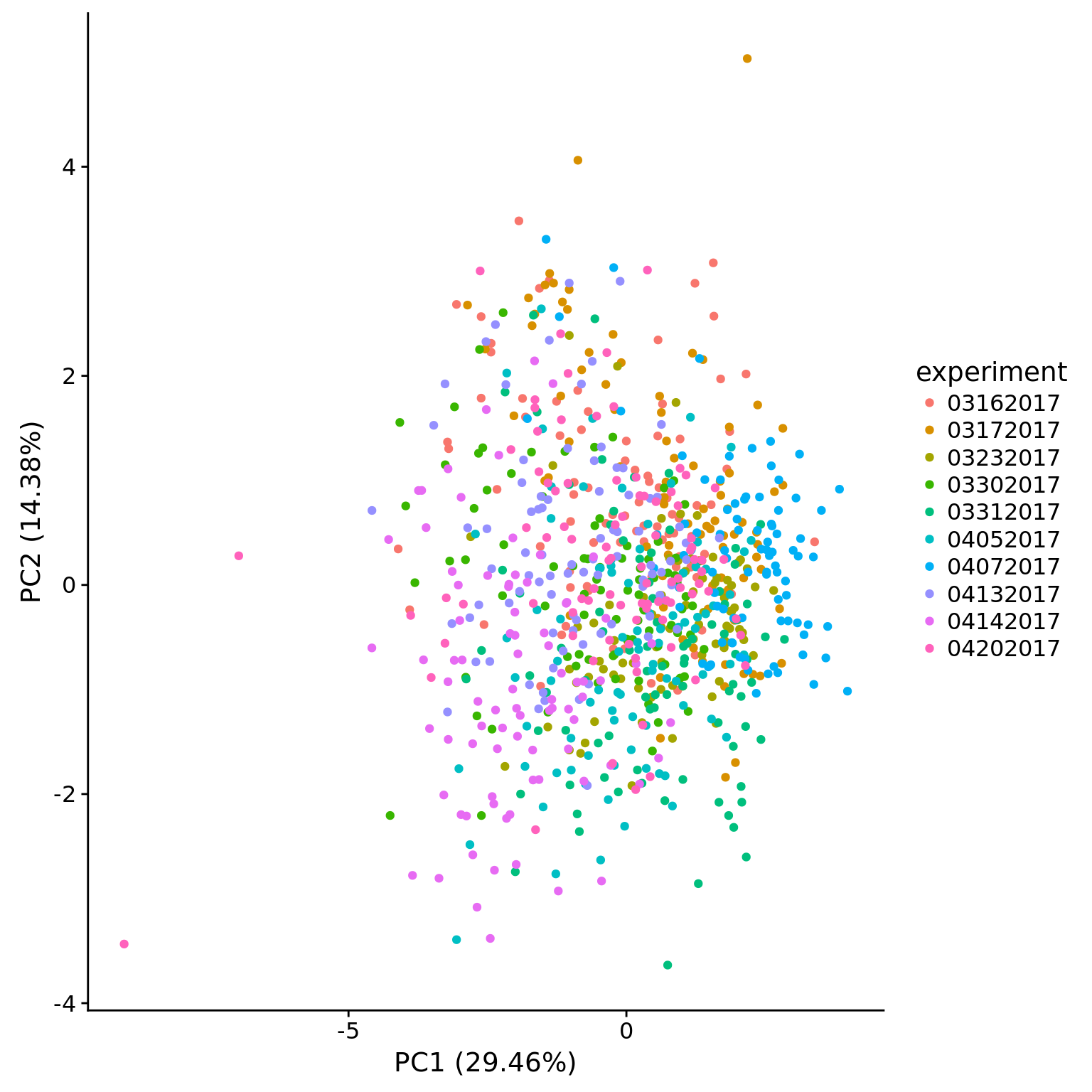
plot_pca(pca_ce$PCs, pcx = 1, pcy = 2, explained = pca_ce$explained,
metadata = pData(eset_ce_clean),
color = "worm", factors = "worm")
Human
Remove zeros.
eset_hs_clean <- eset_hs[rowSums(exprs(eset_hs)) != 0, ]
dim(eset_hs_clean)Features Samples
18904 869 Only keep genes which are observed in at least 50% of the samples.
eset_hs_clean <- eset_hs_clean[apply(exprs(eset_hs_clean), 1, present), ]
dim(eset_hs_clean)Features Samples
6587 869 Convert to log2 counts per million.
mol_hs_cpm <- cpm(exprs(eset_hs_clean), log = TRUE)
hist(rowMeans(mol_hs_cpm))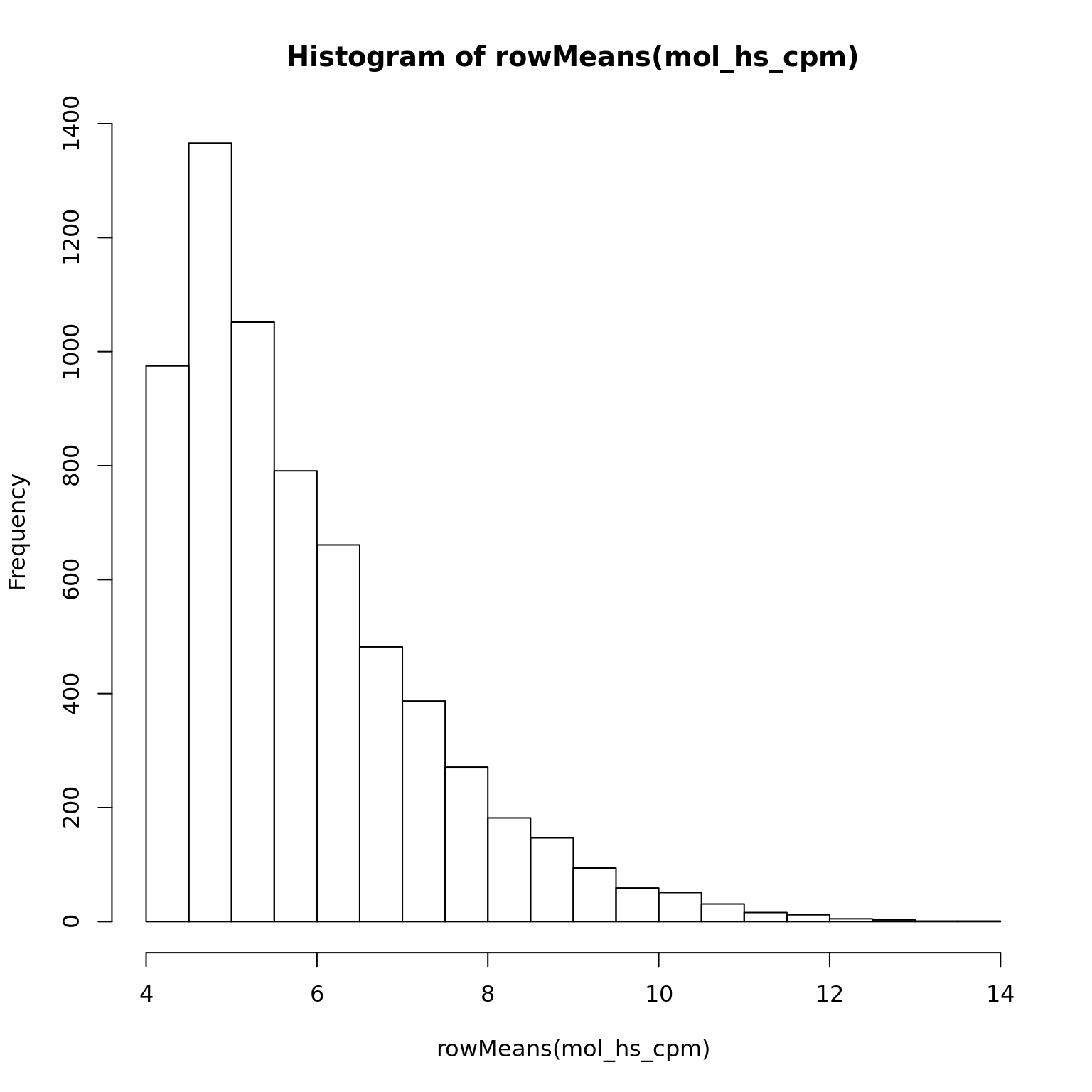
pca_hs <- run_pca(mol_hs_cpm)
plot_pca(pca_hs$PCs, pcx = 1, pcy = 2, explained = pca_hs$explained,
metadata = pData(eset_hs_clean), color = "experiment")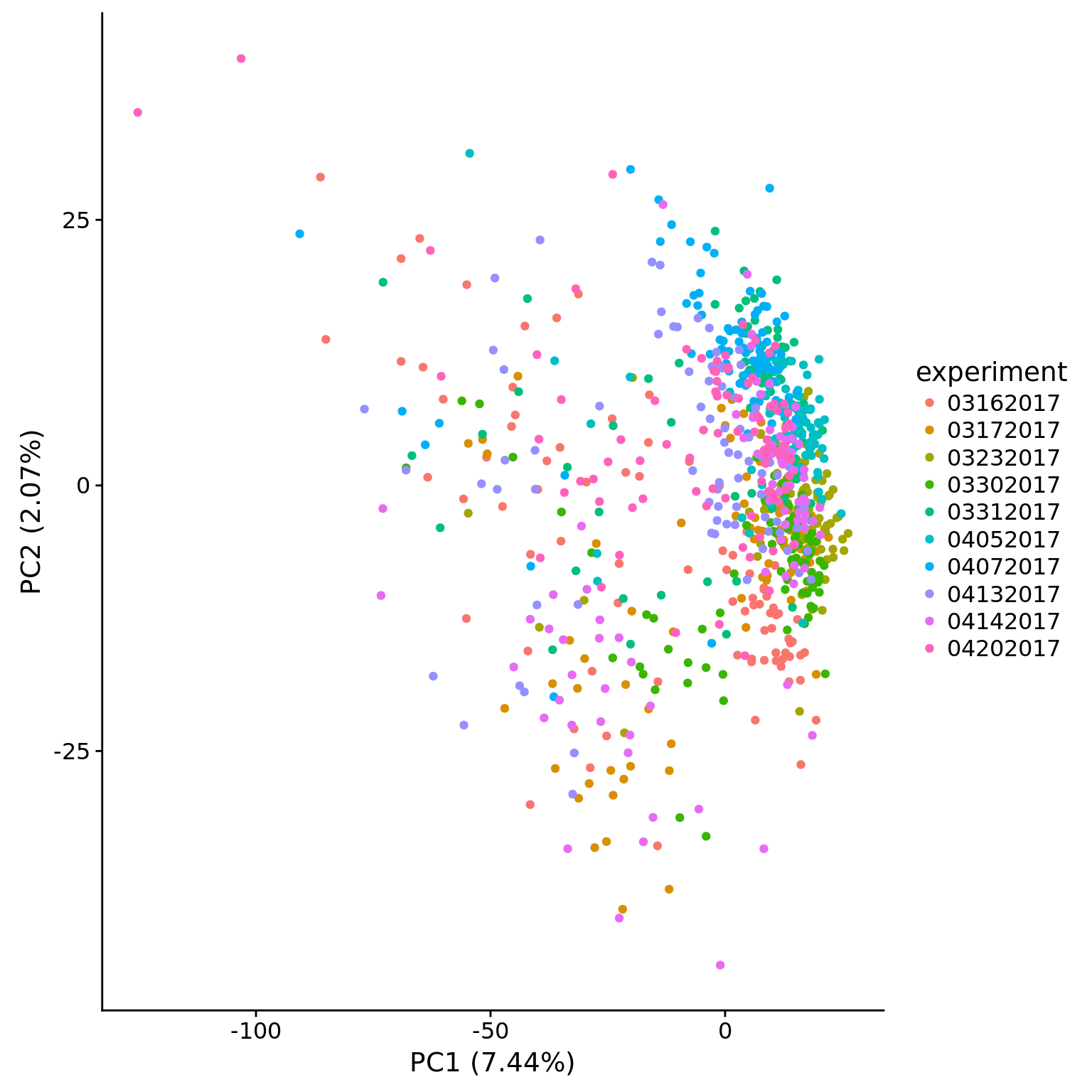
plot_pca(pca_hs$PCs, pcx = 3, pcy = 4, explained = pca_hs$explained,
metadata = pData(eset_hs_clean), color = "experiment")
Visualizing how cells cluster by chip and individual for a subset of individuals.
pca_hs_data <- cbind(pca_hs$PCs[, 1:4], pData(eset_hs_clean))
hs_pc1v2 <- ggplot(pca_hs_data %>% filter(chip_id %in% c("NA18505",
"NA18507",
"NA18508")),
aes(x = PC1, y = PC2, color = experiment)) +
geom_text(aes(label = str_sub(chip_id, 6, 7)))
hs_pc1v2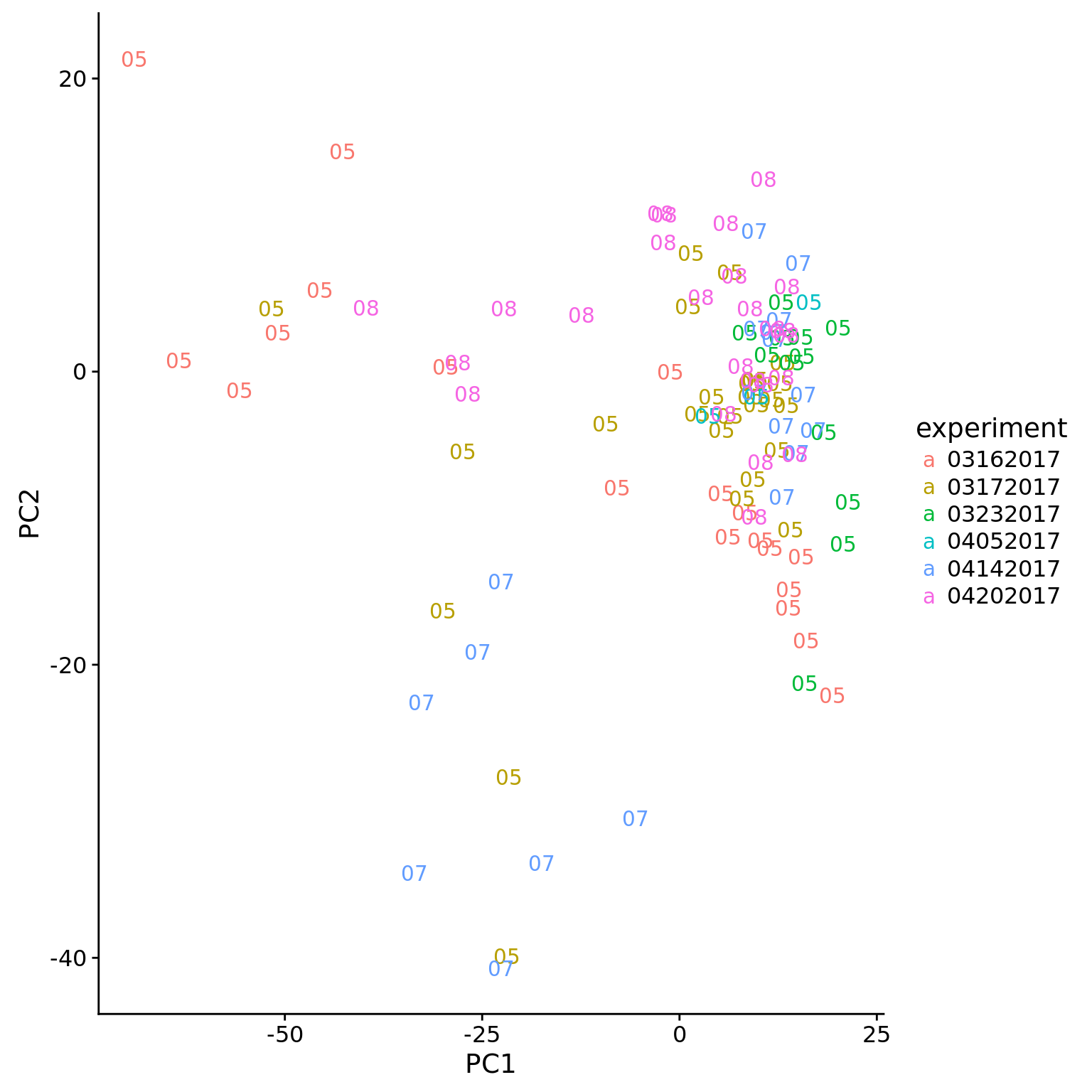
hs_pc3v4 <- hs_pc1v2 %+% aes(x = PC3, y = PC4)
hs_pc3v4
Visualizing C1 chips 04132017 and 04142017 because they share 3 individuals. PC1 and PC2 separate outlier cells. PC3 appears to separate by chip followed by individual in PC4.
pca_cross_chip_1v2 <- ggplot(pca_hs_data %>%
filter(experiment %in% c("04132017", "04142017"),
chip_id %in% c("NA18498", "NA18510", "NA18520",
"NA18522", "NA19203")),
aes(x = PC1, y = PC2, shape = experiment,
color = chip_id)) +
geom_point() +
labs(title = "C1 chips 04132017 & 04142017")
pca_cross_chip_3v4 <- pca_cross_chip_1v2 %+% aes(x = PC3, y = PC4)
plot_grid(pca_cross_chip_1v2, pca_cross_chip_3v4, labels = LETTERS[1:2])
Session information
sessionInfo()R version 3.4.1 (2017-06-30)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Scientific Linux 7.2 (Nitrogen)
Matrix products: default
BLAS: /project2/gilad/jdblischak/miniconda3/envs/scqtl/lib/R/lib/libRblas.so
LAPACK: /project2/gilad/jdblischak/miniconda3/envs/scqtl/lib/R/lib/libRlapack.so
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] parallel methods stats graphics grDevices utils datasets
[8] base
other attached packages:
[1] bindrcpp_0.2 testit_0.6 Biobase_2.38.0
[4] BiocGenerics_0.24.0 tidyr_0.7.1 stringr_1.2.0
[7] edgeR_3.20.1 limma_3.34.1 dplyr_0.7.4
[10] cowplot_0.9.1 ggplot2_2.2.1
loaded via a namespace (and not attached):
[1] Rcpp_0.12.13 compiler_3.4.1 git2r_0.19.0 plyr_1.8.4
[5] bindr_0.1 tools_3.4.1 digest_0.6.12 evaluate_0.10.1
[9] tibble_1.3.3 gtable_0.2.0 lattice_0.20-34 pkgconfig_2.0.1
[13] rlang_0.1.2 yaml_2.1.14 knitr_1.16 locfit_1.5-9.1
[17] rprojroot_1.2 grid_3.4.1 glue_1.1.1 R6_2.2.0
[21] rmarkdown_1.6 purrr_0.2.2 magrittr_1.5 backports_1.0.5
[25] scales_0.5.0 htmltools_0.3.6 assertthat_0.1 colorspace_1.3-2
[29] labeling_0.3 stringi_1.1.2 lazyeval_0.2.0 munsell_0.4.3 This R Markdown site was created with workflowr