Images quality control follow-up analysis
Joyce Hsiao
Last updated: 2017-12-11
Code version: ff5a8ce
Background and summary
In the current analysis, we computed corrected signal for each channel as follows:
(foreground mean intensity - background mean intenstiy)*mask sizeThis is equivalent to computing the sum of pixel intensities after substracting background mean intensity from each pixel in the mask. The mask size is defined as: Denote the center of the nucleus as (cx,cy), then the mask area is (max(1,cx-50):min(cx+50,nrow), max(1,cy-50):min(cy+50,ncol)). The maximum mask size is 10,000.
This metric for summarizing pixel intensity factors into the quantity of cell cycle protein (green and red), and indirectly, the cell size. While, previously our metric of mean pixel intensity does not account for the quantity of cell cycle protein (green and red).
Results:
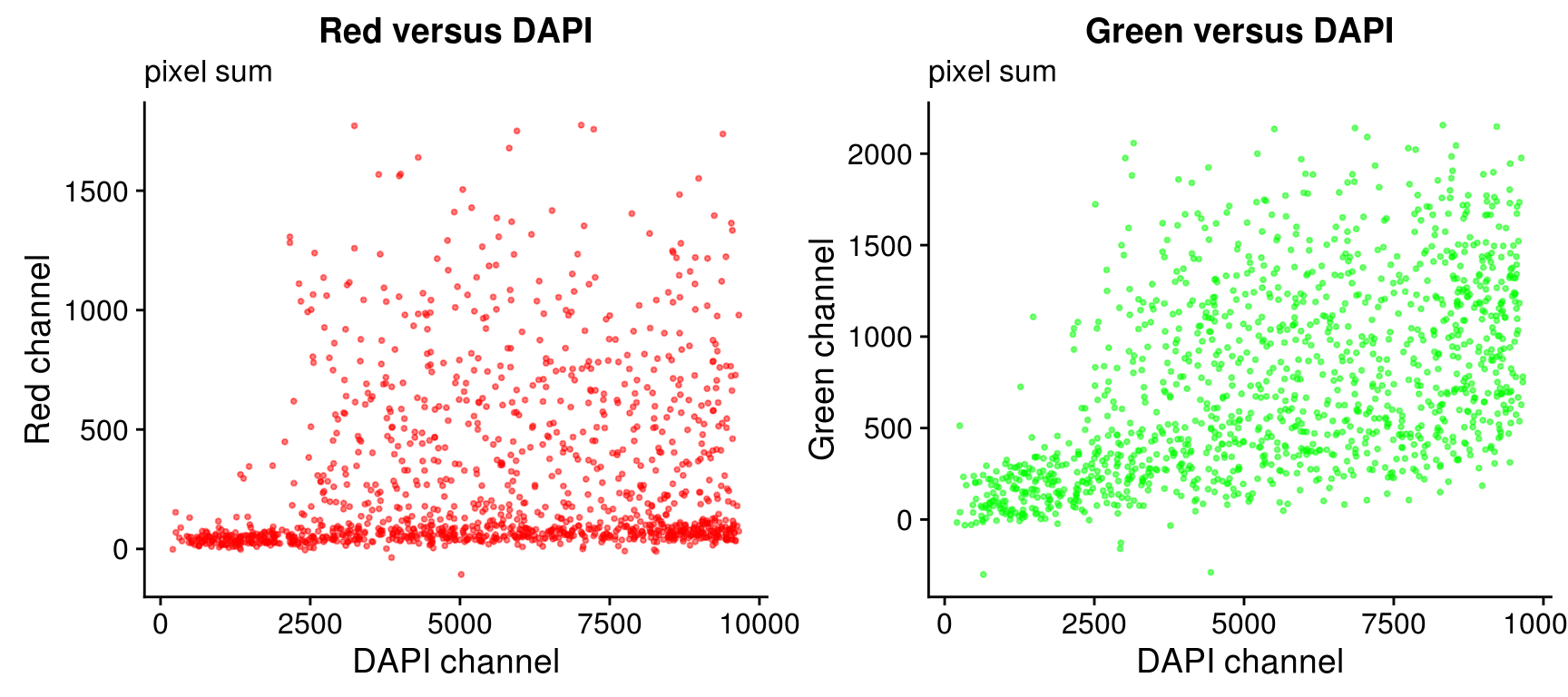
In about 10 to 15 single cell samples, the background intensity is greater than the foreground intensity by a small amount (~.001) in the Red or the Green channel. we need to check to see if there are signals at all for these samples in any of channles. If there’s any signal, then we’ll set the channel with no signal to the lowest possible intensity value.
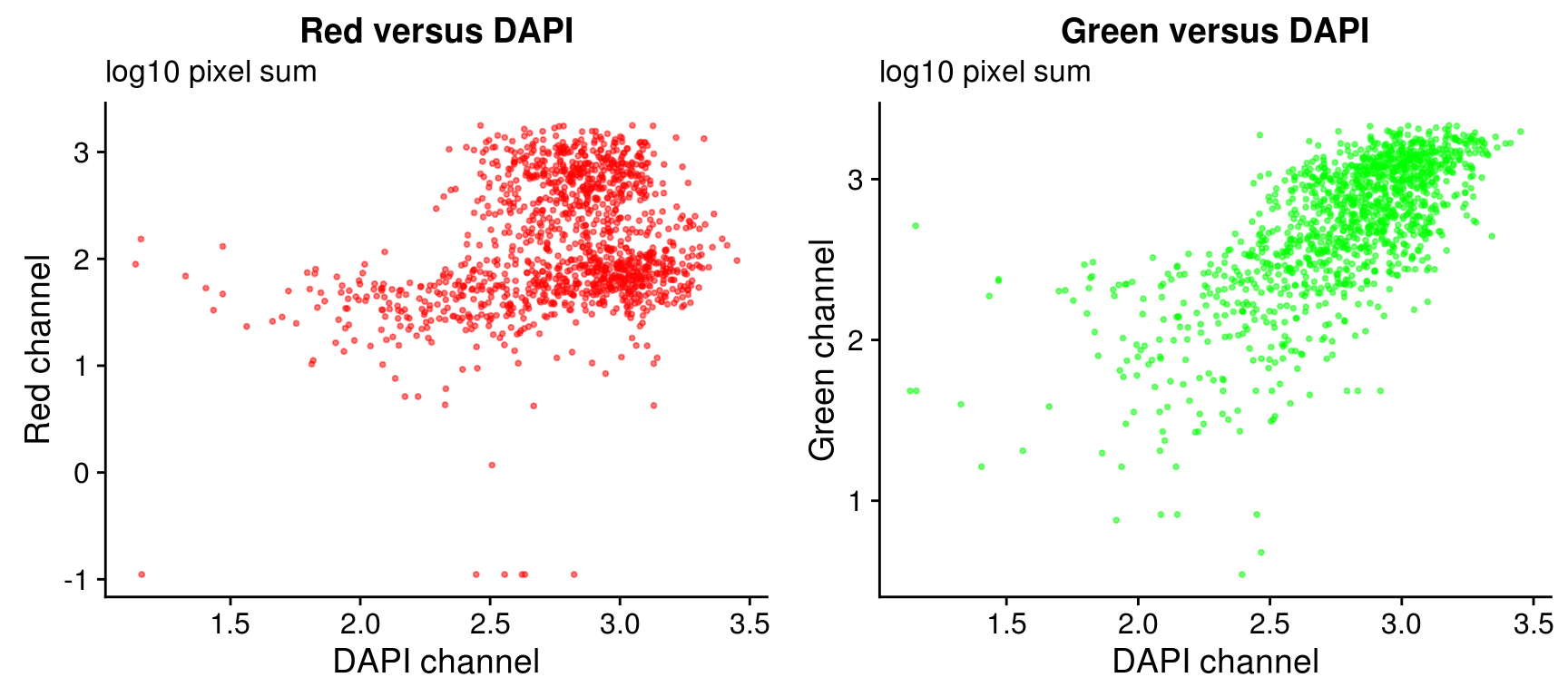
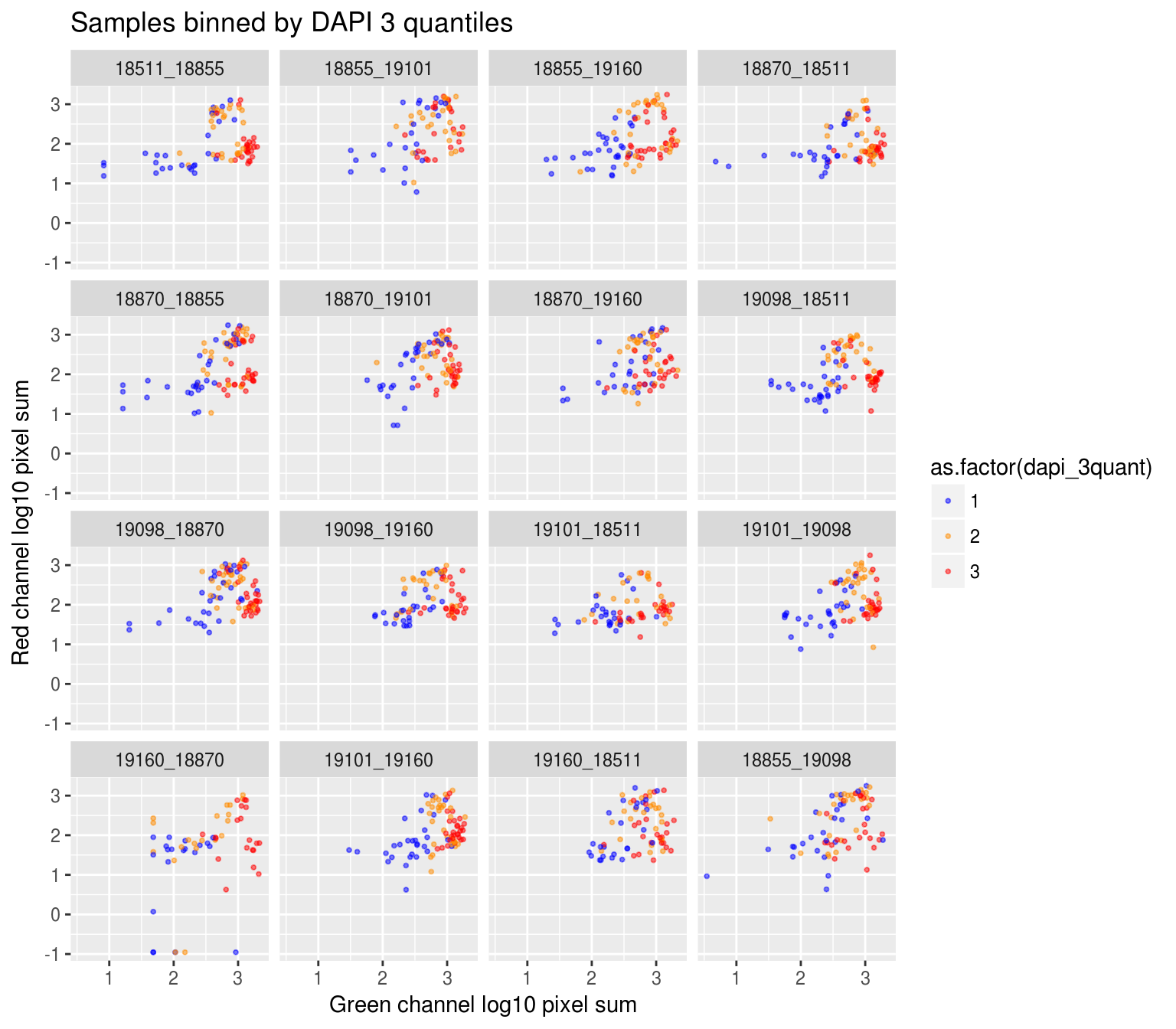
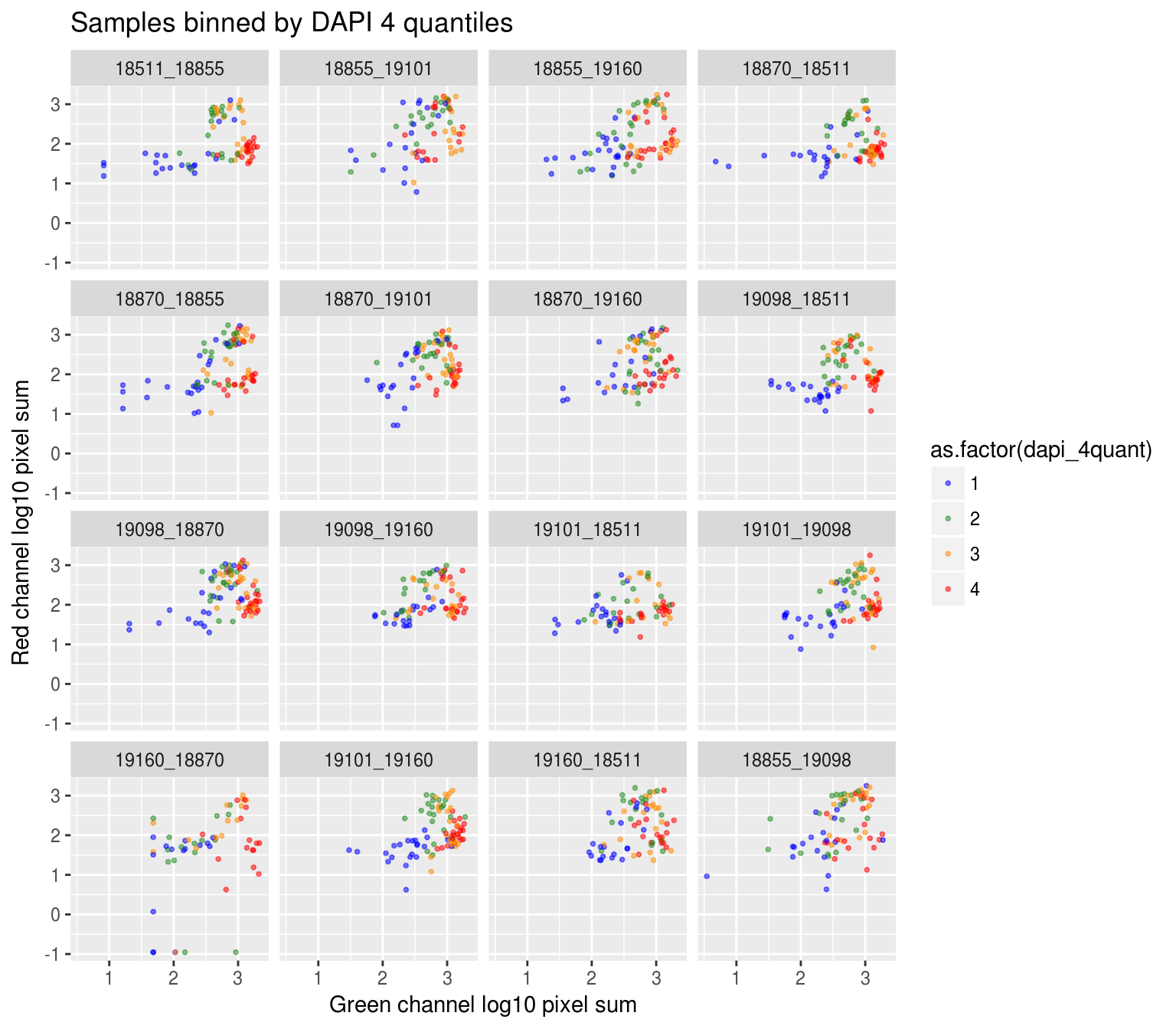
On log10 scale of pixel sum intensity, we are able to observe the separation of single cell samples on Red channel and also on Green channel. And, interestly, in figures plotting Red against Green, we observe three clusters roughly correspond to low, medium and high DAPI.
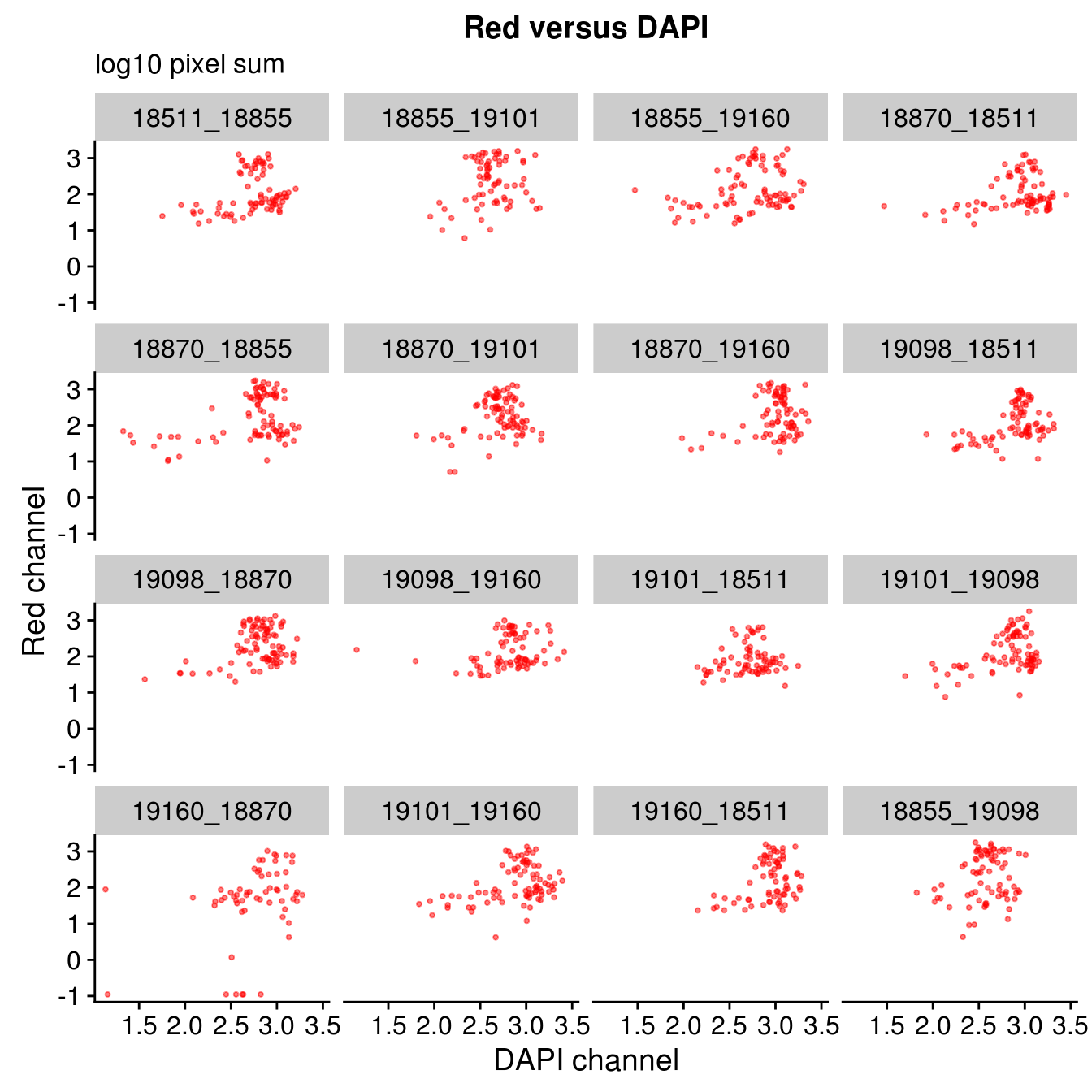
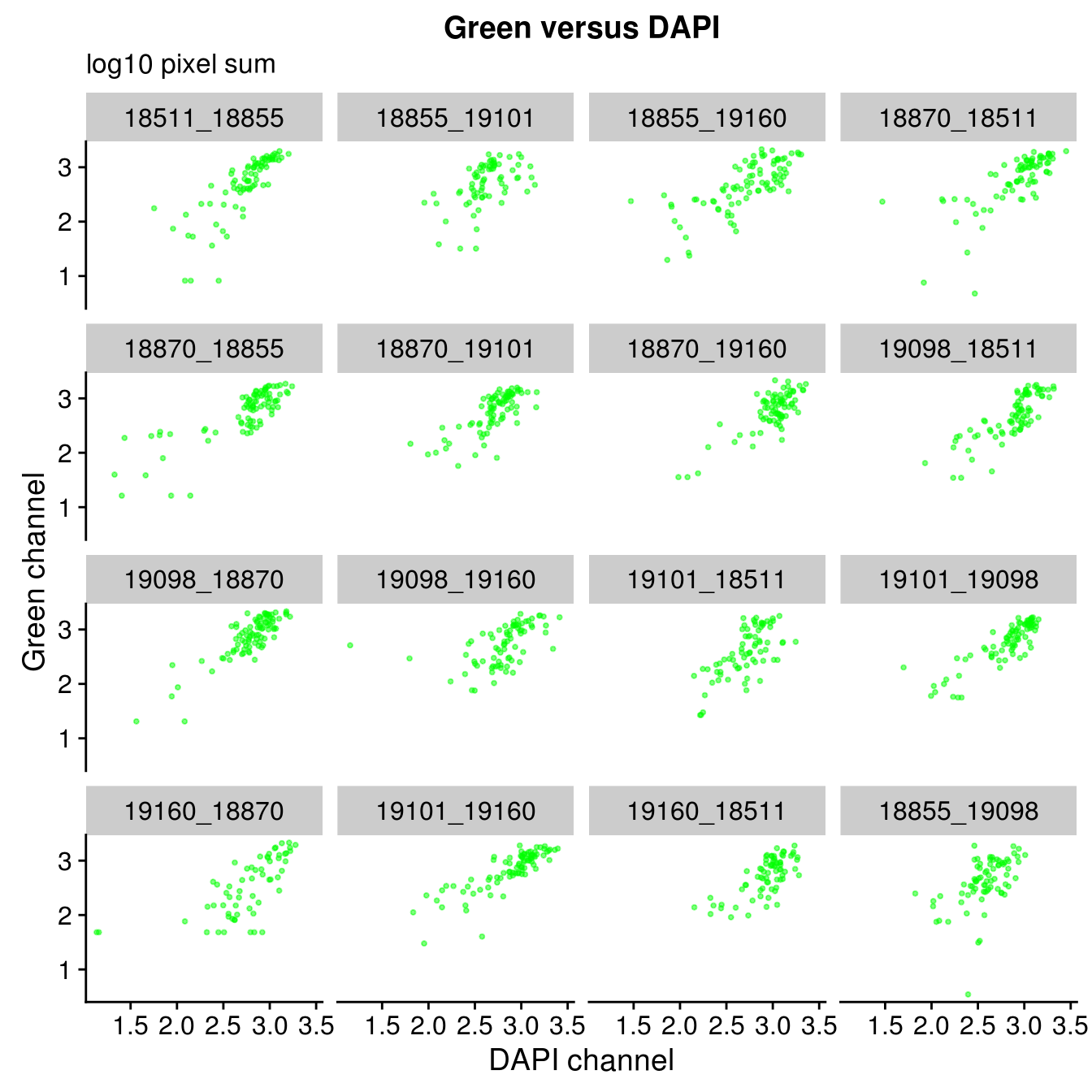
Plate effect: the pattern observed in Red intensities against Green intensities appears to vary by plate, and when we control for plate differencees in DAPI intensities, we see the distribution of DAPI intensities fairly similar across plates!
The metric for intensity in the current analysis effective scales the mean intensity by the channel specific and sample-specific coverage area of fluorescent intensities. This is likely the main contributing factor for the better separtion of single cell samples we observe here.
Next steps:
Batch correction for plate and individual effect.
Classification? How? Should be non-linear…
Loading data
Warning: Installed Rcpp (0.12.14) different from Rcpp used to build dplyr (0.12.10).
Please reinstall dplyr to avoid random crashes or undefined behavior.Import combined intensity data (see combine-intensity-data.R).
ints <- readRDS(file="/project2/gilad/joycehsiao/fucci-seq/data/intensity.rds")
# compute sum of intensities
ints$dapi.sum.zoom <- with(ints, (dapi.fore.zoom.mean-dapi.back.zoom.mean)*10000)
ints$rfp.sum.zoom <- with(ints, (rfp.fore.zoom.mean-rfp.back.zoom.mean)*10000)
ints$gfp.sum.zoom <- with(ints, (gfp.fore.zoom.mean-gfp.back.zoom.mean)*10000)DAPI versus Green/Red
Pixel sum
log 10 pixel sum
By plate
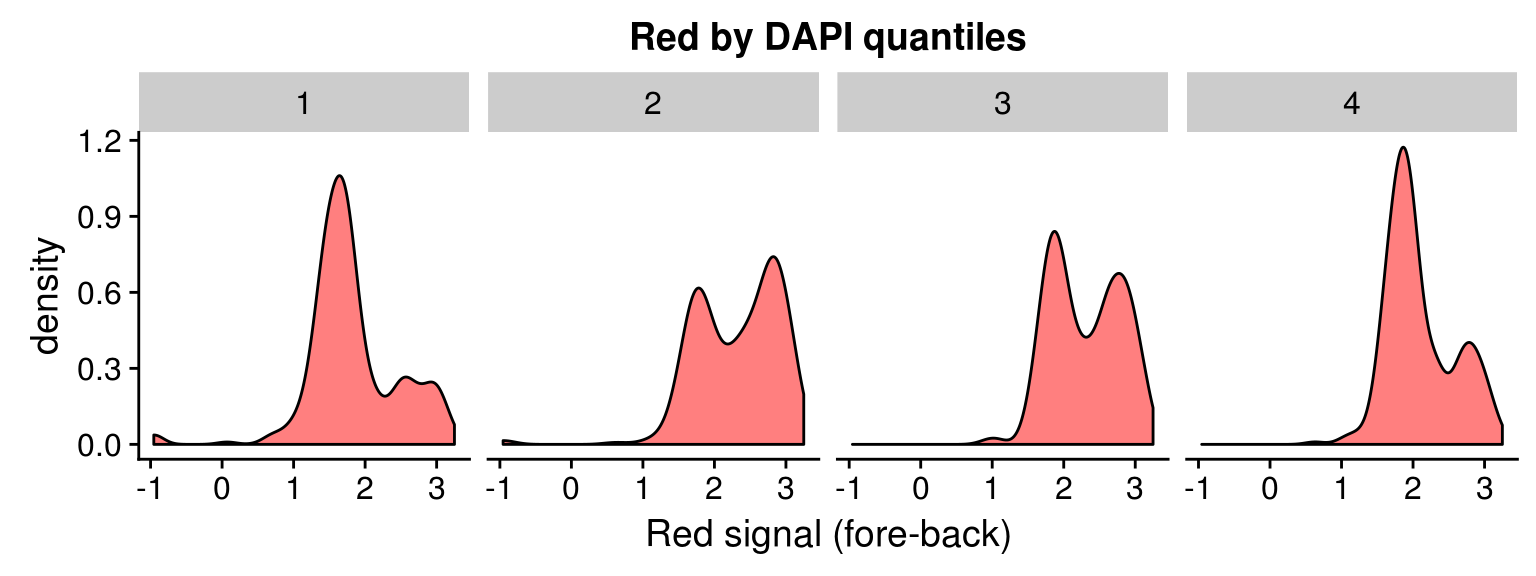
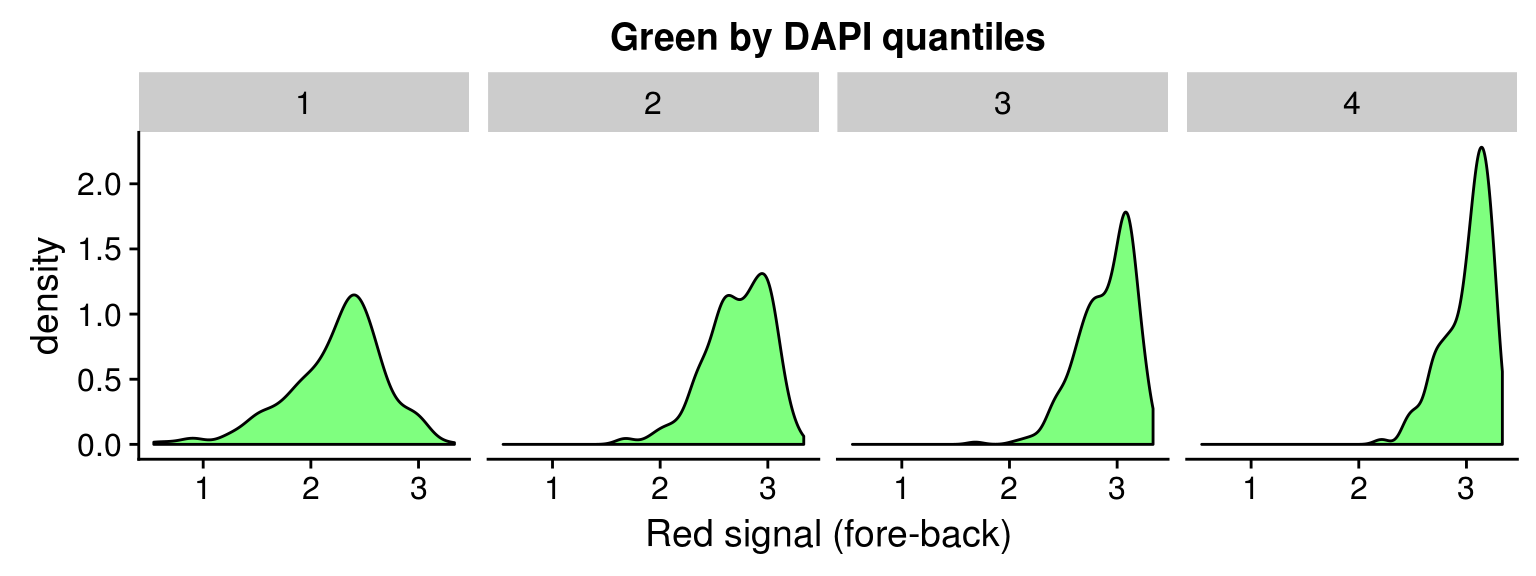
Red vs Green binned by DAPI quantiles
Across plates
Compute DAPI quantiles across plates.
ints <- ints %>% mutate(dapi_4quant=ntile(dapi.mean.log10sum,4),
dapi_3quant=ntile(dapi.mean.log10sum,3))\(~\)
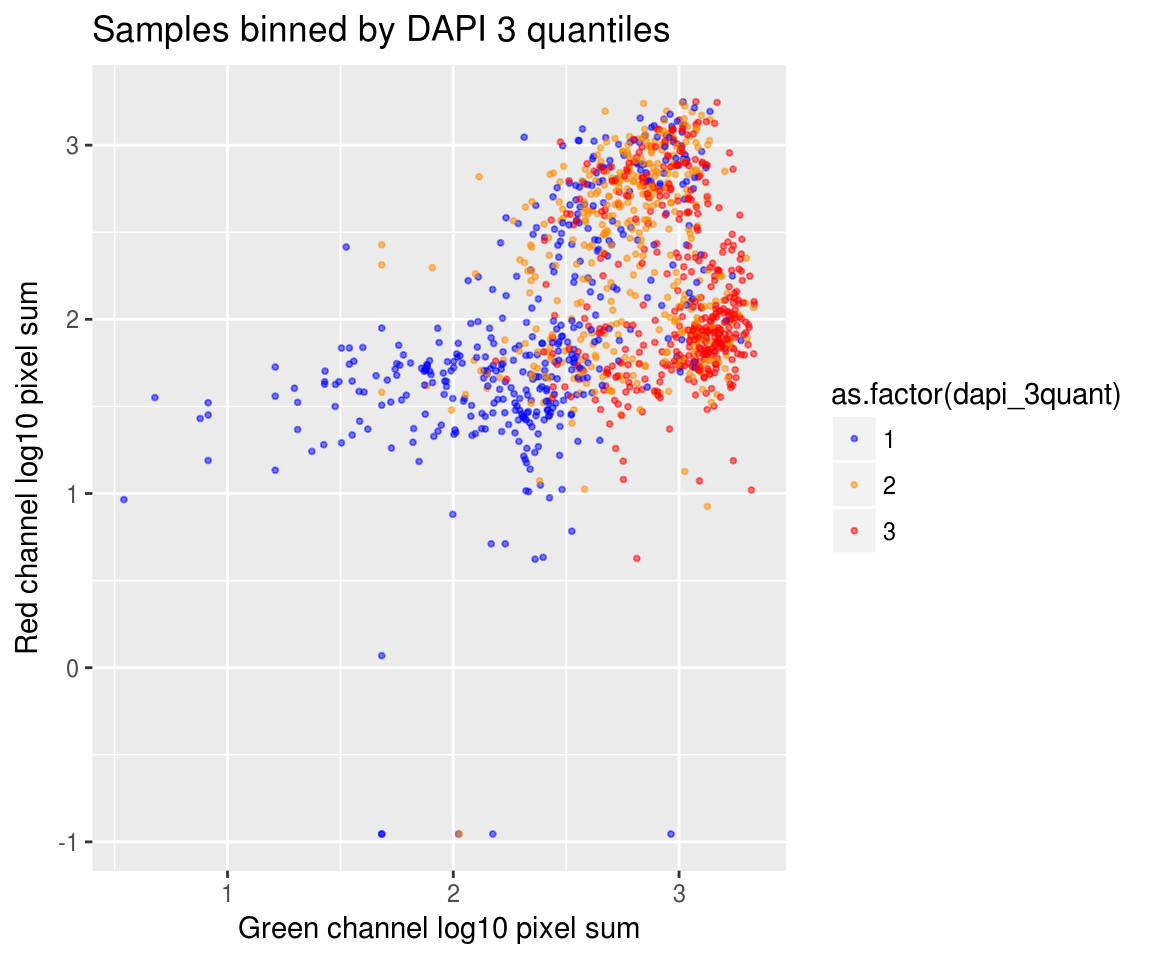
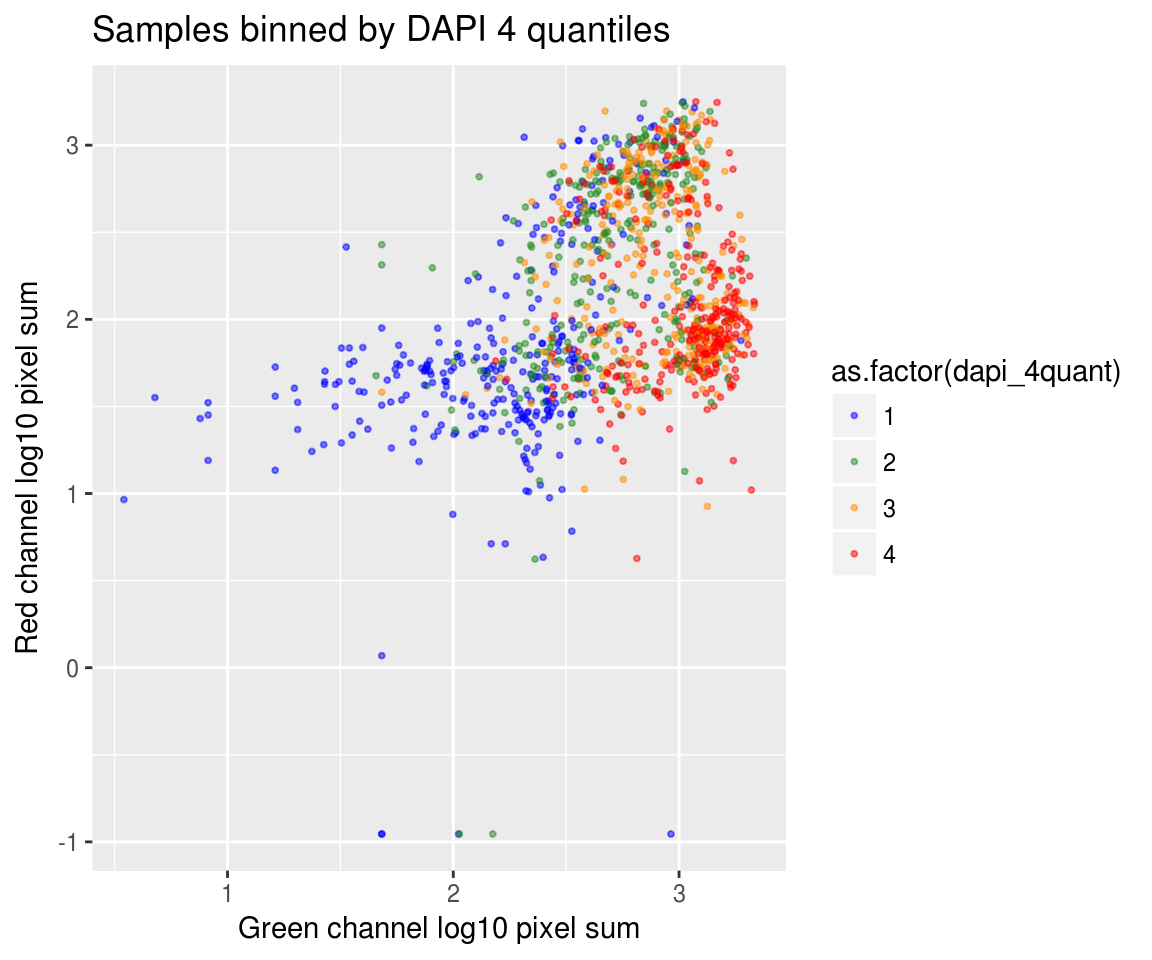
By plate, overall DAPI quantiles
Compute DAPI quantiles across plates.
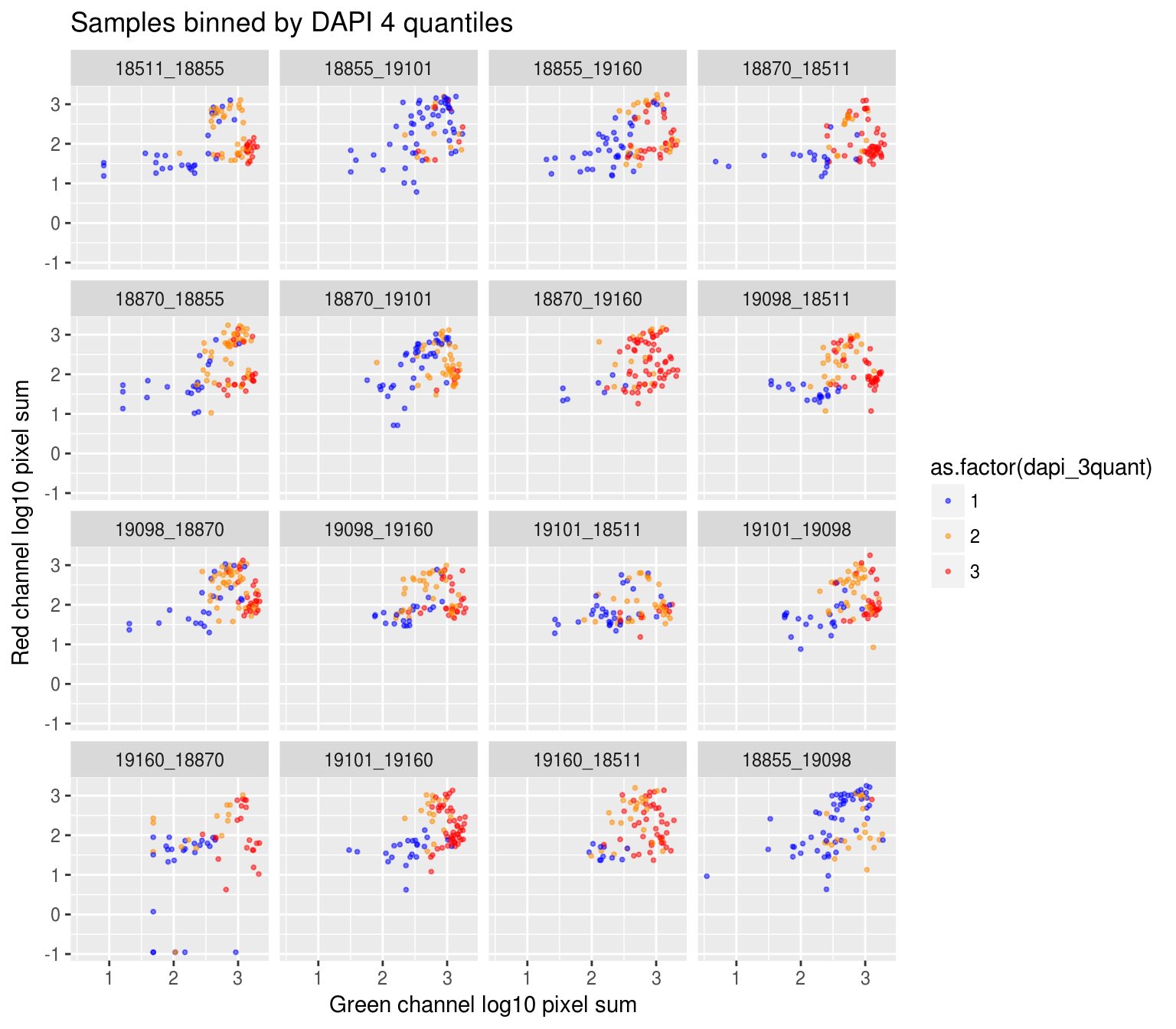
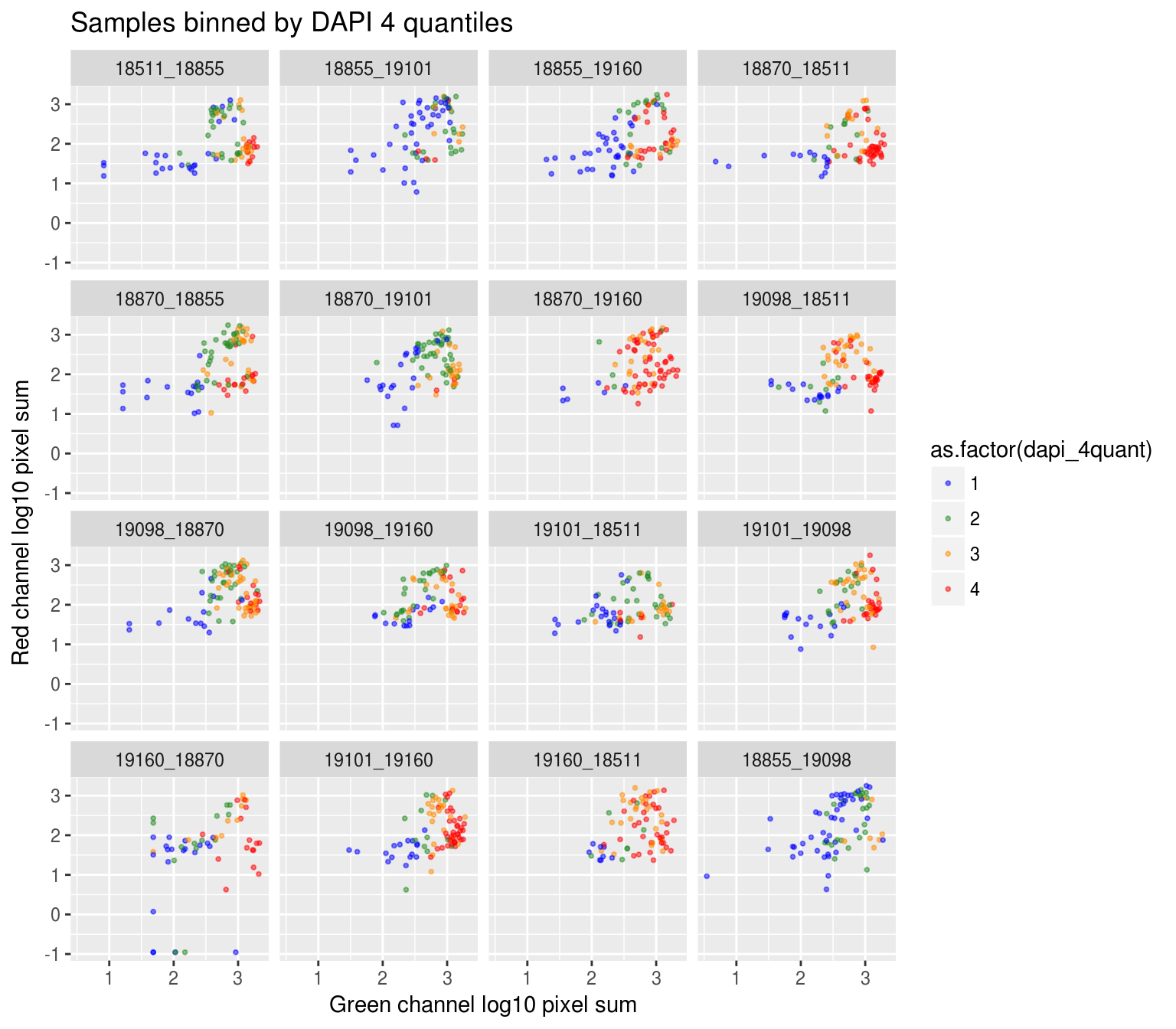
By plate, plate-specific DAPI quantiles
Compute DAPI quantiles in each plate.
ints_tmp <- ints %>% group_by(plate) %>% mutate(dapi_4quant=ntile(dapi.mean.log10sum,4),
dapi_3quant=ntile(dapi.mean.log10sum,3))\(~\)
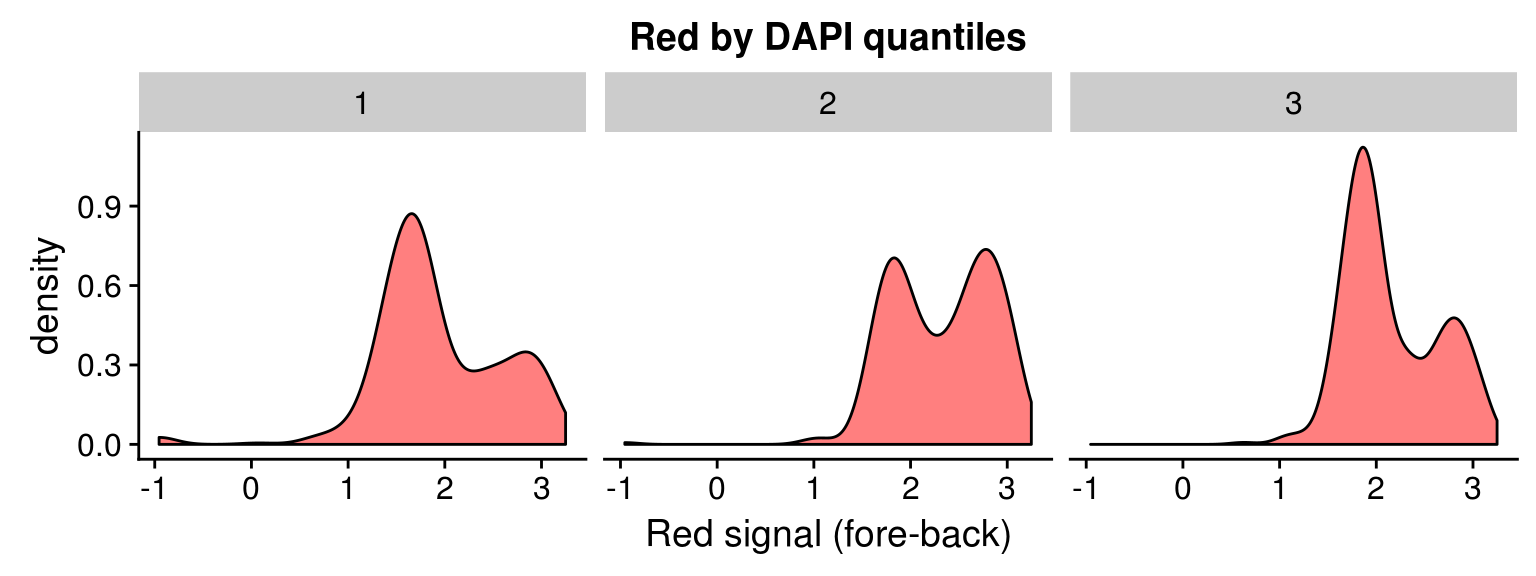
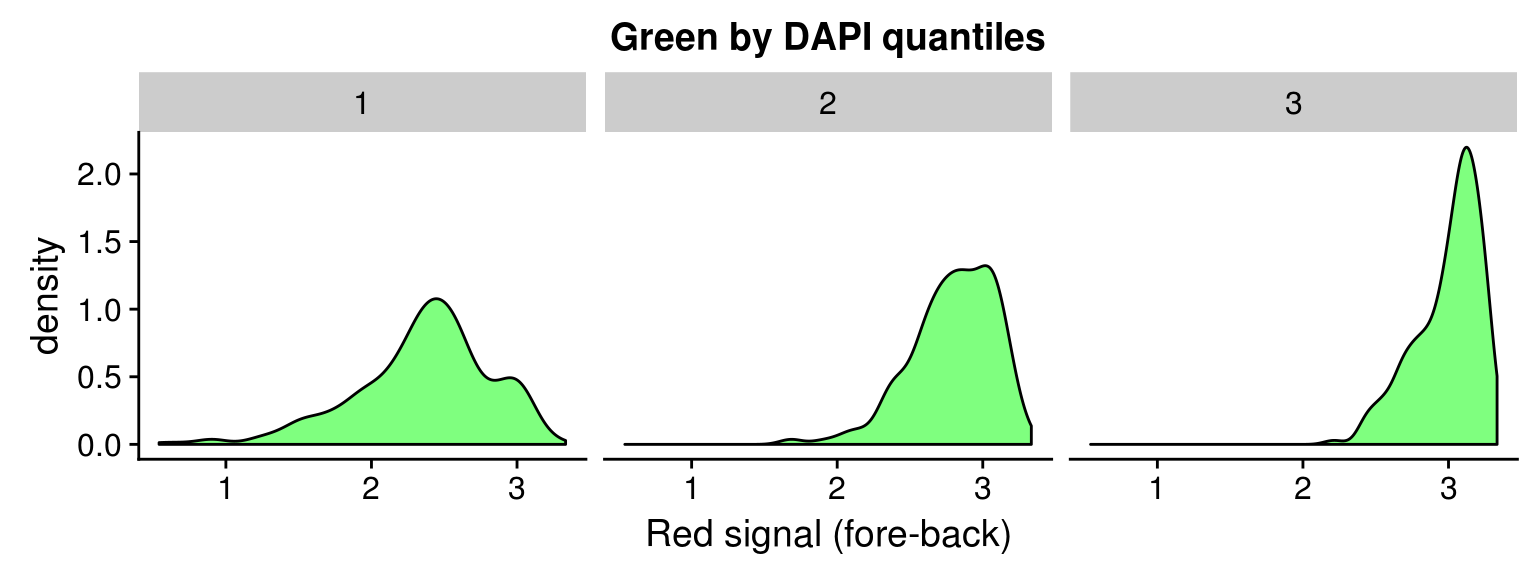
Density distribution
DAPI 3 quantiles across plates
DAPI 4 quantiles across plates
Session information
sessionInfo()R version 3.4.1 (2017-06-30)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Scientific Linux 7.2 (Nitrogen)
Matrix products: default
BLAS: /home/joycehsiao/miniconda3/envs/fucci-seq/lib/R/lib/libRblas.so
LAPACK: /home/joycehsiao/miniconda3/envs/fucci-seq/lib/R/lib/libRlapack.so
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] RColorBrewer_1.1-2 wesanderson_0.3.2 cowplot_0.8.0
[4] ggplot2_2.2.1 dplyr_0.7.0
loaded via a namespace (and not attached):
[1] Rcpp_0.12.14 knitr_1.16 magrittr_1.5 munsell_0.4.3
[5] colorspace_1.3-2 R6_2.2.0 rlang_0.1.2 stringr_1.2.0
[9] plyr_1.8.4 tools_3.4.1 grid_3.4.1 gtable_0.2.0
[13] git2r_0.19.0 htmltools_0.3.6 yaml_2.1.14 lazyeval_0.2.0
[17] rprojroot_1.2 digest_0.6.12 assertthat_0.1 tibble_1.3.3
[21] glue_1.1.1 evaluate_0.10.1 rmarkdown_1.6 labeling_0.3
[25] stringi_1.1.2 compiler_3.4.1 scales_0.4.1 backports_1.0.5 This R Markdown site was created with workflowr